
Синдром жаберных дуг и рождение хдорового ребенка
Содержание
Синдром Гольденхара: симптомы, выбор лечения и прогноз течения патологии
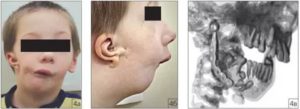
Болезнь проявляется комплексом симптомов, основным из которых является односторонняя дисплазия черепа, аномалии развития ушных раковин, зубов, век, позвоночника.
Синдром Гольденхара – это редкая форма генетического заболевания, причиной которого является нарушение развития первой и второй жаберных дуг.
Зная, как распознать болезнь на ранней стадии, вы сможете добиться при лечении эффективных результатов.
Проявления синдрома
Гипоплазия половины лица и его асимметрия. Одна половина лица меньше, с недоразвитыми верхней и нижней челюстями и мимическими мышцами.
- Низкорасположенное, недоразвитое ухо, с узким наружным слуховым проходом, дефектами барабанной перепонки. В некоторых случаях ухо может отсутствовать. Отмечается проводниковая тугоухость или глухота.
- Сенсоневральная глухота.
- Недоразвитое мягкое небо, губы, гланды. Трахеопищеводный свищ. Рот больше средних размеров, при этом один угол рта выше другого.
- Колобома верхнего века. Страбизм. Дермоидная киста. Недоразвитый глаз или полное его отсутствие.
- Дефекты шейных позвонков: недоразвитость позвонков, сращение позвонков, короткая шея.
- Дефекты грудных позвонков и ребер: отсутствие некоторых ребер или лишние ребра, сколиоз.
- Дефекты сердечно-сосудистой системы: открытый артериальный проток, отверстия в межжелудочковой перегородке, тетрада Фалло, коарктация аорты, стеноз легочной артерии.
- Гипоплазия легких, вызванная грубыми дефектами развития грудной клетки.
- Агенезия (отсутствие) почек, гидронефроз, мультикистоз. Недоразвитость или отсутствие матки.
- Нарушения со стороны центральной нервной системы: трудности с обучением, которые могут быть вызваны как глухотой, так и структурными изменениями (гидроцефалия, мальформация Арнольда–Киари, затылочное энцефалоцеле, агенезия мозолистого тела, гипоплазия прозрачной перегородки). Парез лицевого нерва.
Причины
Не установлено точно, какие имеет синдром Гольденхара причины, но большинство ученых придерживаются мнения относительно генетической природы заболевания.
Эпизоды заболевания имеют случайный характер, но нередко после расспросов родственников пациентов прослеживается фактор наследственности.
Некоторые теории рассматривают связь развития патологии с воздействием на ранних сроках беременности некоторых химических веществ, вирусных патогенов.Кроме того, факторами риска развития недуга считаются следующие факты из анамнеза беременной:
- кровнородственный брак;
- предшествующие прерывания беременности;
- чрезмерная масса тела;
- сахарный диабет.
Диагностика
Предварительный диагноз патологии устанавливают сразу после рождения ребенка на основании визуального осмотра. Диагностика включает тщательный опрос родителей и составление анамнеза.
Разнообразные диагностические процедуры позволяют поставить точный диагноз.
- Определение остроты слуха — регистрация активности слухового нерва после короткой акустической стимуляции, аудиометрия, импедансометрия.
- Осмотр глаза и проведение офтальмологического исследования.
- Электрокардиография.
- Ренгенография черепа, шеи, грудной клетки.
- УЗИ внутренних органов.
- КТ головного мозга.
- Медико-генетическое консультирование.
Лечение синдрома Гольденхара
Развитие патологий черепа и позвоночника, а также других систем органов с этим синдромом требует адекватного лечения специалистов различных профилей. Больной с лёгкой формой заболевания наблюдается до 3 лет, после этого показано хирургическое лечение.
При тяжёлой форме наследственного заболевания Гольденхара применяют хирургическое лечение (в раннем возрасте и по достижении 2 лет). Затем проводится комплексное устранение отдельных симптомов заболевания.
Однако стоит отметить, что данное заболевание без поэтапной хирургической терапии вылечить невозможно. Степень и тяжесть выявленных пороков определяют, сколько хирургических вмешательств необходимо провести. В чем заключается хирургическое вмешательство?
Таким больным обычно проводят:
- компрессионно-дистракционную репозицию костных отломков;
- замену повреждённого височно-нижнечелюстного сустава, нижней и верхней челюсти;
- остеотомию носа, нижней и верхней челюстей, корректирующую аномалию их развития;
- пластическую коррекцию подбородка и носа.
Препараты
Для лечения возможных воспалительных процессов и ускорения реабилитационного процесса назначается курс антибиотиков и витаминов. При челюстно-лицевых хирургических вмешательствах назначается остеотропные препараты: эритромицины, пенициллины, линкомицины.
Пациенту с синдромом Гольденхара до и после операции врачом назначаются анальгезирующие препараты. Малышам прописывают детский «Нурофен», быстро снимающий болевые ощущения, температуру и воспалительные процессы, который обладает пролонгированным эффектом (до девяти часов).
Дозировка средства достигает тридцати мг на килограмм веса ребенка ежедневно.
В период реабилитации больному необходимо полноценно питаться и пройти витаминную терапию. Витамины в обязательном порядке должны содержать витамины группы D и B, аскорбиновую кислоту, ретинол, токоферол и др.
Для того чтобы избежать инфекций и ускорить рассасывание послеоперационных отеков и швов, необходимо пройти физиопроцедуры ультрафиолетовым излучением, ультразвуковую терапию и электромагнитотерапию, также лазеро- и магнитотерапию, а также в комплексе, гипербарическую оксигенацию.
Ортодонтическая терапия подразумевает профилактику аномального развития челюстей, исправление патологического прикуса, подготовку зубов и мимической мускулатуры к операциям.
Прогноз
Для пациентов с синдромом Гольденхара прогноз в большинстве случаев благоприятный, при этом многое зависит от масштабов поражения внутренних органов. При выявлении всего комплекса аномалий, применении всех возможных методик по коррекции нарушений, внимательном отношении близких к пациенту, психологической поддержке есть шанс на полное выздоровление.
(1 5,00 из 5)
Загрузка…
Источник: https://osindromah.ru/geneticheskie/sindrom-goldenhara.html
Синдром Гольденхара: что это, причины, симптомы, лечение и операция

Синдром Гольденхара — наследственная односторонняя гипоплазия лица с преимущественным поражением ушной раковины, глаз, носа, зубов, позвоночника.
При этом вторая половина лица имеет абсолютно нормальный вид.
Это редко встречающееся, генетически обусловленное заболевание отличается неполноценным развитием скелета, мышц, сосудисто-нервных стволов и прочих структур человеческого организма.
Окуло-аурикуло-вертебральная дисплазия — второе название данного недуга. Синдром Гольденхара представляет собой отдельный вид целой группы патологий, для которой характерна задержка физического развития человека. Непосредственной причиной его развития является внутриутробное повреждение жаберных дуг, из которых у плода формируются структуры слухового и жевательного аппарата.
Синдром Гольденхара отличается широким клиническим полиморфизмом. Классическими симптомами заболевания являются: недоразвитие в процессе эмбриогенеза нижней челюсти, резкое нарушение симметрии лица, деформация наружного уха и глаза, нарушение строения позвонков шеи.
Возможно полное отсутствие ушной раковины или органа зрения. У больных формируются дермоидные кисты в глазах, развиваются отклонения в работе сердечно-сосудистой, мочеполовой систем и желудочно-кишечного тракта.
Клинически это проявляется одышкой, нарушением процесса питания, снижением слуха с пораженной стороны. Синдром Гольденхара не связан с умственной отсталостью.
Выявить патологию можно на 20-24 неделе беременности с помощью ультразвуковой диагностики со сканированием в трех измерениях. Большинство случаев синдрома – спорадические.
Впервые патологию описал в 1952 году американский доктор Гольденхар. Синдром получил свое название по фамилии его первооткрывателя. У мальчиков заболевание встречается несколько чаще, чем у девочек. С помощью современных методов диагностики заболевание обнаруживают внутриутробно и решают вопрос о целесообразности сохранения беременности.
Этиология и патогенез
Причины возникновения и механизмы развития синдрома Гольденхара остаются неизвестными. Мнения ученых сходятся на том, что патология относится к наследственным недугам. У пациентов с данным синдромом обнаруживают хромосомные аномалии и мутации генов. Больной ребенок рождается с множественными дефектами лица и патологиями внутренних органов.
Факторы риска рождения больных детей:
- кровнородственный брак,
- частые аборты у матери,
- тяжелые эндокринопатии,
- лишний вес матери,
- воздействие химических веществ и патогенных биологических агентов на ранних сроках беременности.
Во время замены источника кровоснабжения на участке 1 и 2 жаберных щелей эмбриона происходит кровоизлияние. Разрыв кровеносного сосуда в этом месте приводит к нарушению митотического деления клеток и неправильному формированию основных анатомических структур человеческого организма.
Симптоматика
Первые симптомы патологии можно заметить сразу после рождения ребенка. У больных детей асимметричное лицо, мелкие глазницы, деформированные ушные раковины, недоразвитая нижняя челюсть. Это внешние признаки патологии, которые нередко сопровождаются патологическими изменениями во внутренних органах.
Симптомы со стороны зрительного анализатора:
- Анофтальмия,
- Микрофтальм,
- Микрокорнеа — малая роговица,
- Аниридия — отсутствие радужки,
- Птоз верхнего века,
- Страбизм – отклонение зрительной оси глаза от точки фиксации,
- Помутнение хрусталика,
- Дермоиды и липодермоиды на поверхности глазного яблока – небольшие опухоли,
- Поражение глазодвигательных мышц.
Клинические признаки при поражении ушей:
- Аномальное расположение ушных раковин,
- Минимальные размеры ушей,
- Деформация ушных раковин,
- Плоская или выступающая форма ушей,
- Характерные выросты на ушных раковинах,
- Дефекты барабанной перепонки,
- Соединение ушной раковины с уголком рта, придающее лицу перекошенный вид,
- Сужение слухового прохода,
- Фистулы и околоушные свищи,
- Отсутствие наружного слухового прохода,
- Снижение слуха,
- Двусторонняя асимметричная кондуктивная тугоухость,
- Полное отсутствие слуха.
Нижняя челюсть у больных недоразвита. Одна половина лица меньше другой, мимические мышцы развиты слабо. Небо имеет вид высокой арки, иногда расщепляется.
Ротовая щель при этом слишком широкая, прикус нарушается, появляются добавочные уздечки и расщелины на малом язычке. Нарушается рост зубов. Мягкое небо, губы и гланды деформированы. Имеются трахеопищеводные свищи.
Рот большой, один угол выше другого. Недоразвиты скулы, лоб выступает, присутствуют аномалии языка.
У деток с синдромом Гольденхара имеются проблемы с позвоночником: недоразвитие шейного отдела, сколиоз, косолапость, клиновидная форма позвонков, их слияние, наличие полупозвонков, сращение шейных позвонков с затылком, костные аномалии, короткая шея, искривление позвоночника.Проявлениями со стороны сердечно-сосудистой системы являются симптомы врожденных пороков сердца. Больные дети рождаются с недоразвитием легких, отсутствием органов малого таза, парезом лицевого нерва, гидроцефалией. Они испытывают трудности в обучении. Каждый 10 больной ребенок рождается с поражением ЦНС.
Для синдрома Гольденхара характерно появление симптоматики с одной стороны лица и туловища.
Осложнения и последствия
тяжелое поражение лица и глаз при СГ
Некоторые проявления синдрома Гольденхара не совместимы с жизнью. Ребенок может умереть сразу после рождения.
Осложнения синдрома Гольденхара:
- неправильный прикус вызывает ряд неудобств,
- развивается гипоплазия лицевых костей,
- трудности с глотанием и жеванием,
- прогрессирующая патология зрения,
- серьезные физические неудобства,
- психологическая травма.
Лечение
Лечение синдрома Гольденхара мультидисциплинарное. Дети до трехлетнего возраста наблюдаются у различных специалистов — ЛОР-врача, окулиста, сурдолога, ортопеда и прочих. Детей старше 3 лет направляют к хирургу. В тяжелых случаях для удаления грубых врожденных дефектов сразу после рождения проводят оперативное лечение, а затем комплексную медикаментозную и ортодонтическую терапию.
Хирургическое лечение
Вид оперативного вмешательства определяется тяжестью патологии.
- После искусственного перелома кости соединяют костные фрагменты и устраняют их подвижность с помощью компрессионно-дистракционных аппаратов.
- Проводят протезирование и реконструкцию височно-нижнечелюстного сустава.
- Устраняют деформации носа и улучшают его функции путем искусственного перелома носовой кости.
- Исправляют патологический прикус оперативным путем.
- Проводят пластические операции — пластику носа, ушных раковин, нижней челюсти.
Чтобы предотвратить развитие воспалительных осложнений, во время реабилитации назначают антибиотики и витамины. К остеотропным антибактериальным препаратам относятся «Линкомицин», «Эритромицин», защищенные пенициллины – «Амоксиклав». Для снятия боли назначают анальгетики – «Нурофен», «Ибуклин». Больные дети должны полноценно питаться и принимать поливитаминные комплексы.
Физиотерапия – неотъемлемый компонент лечебного и реабилитационного процесса любых заболеваний. Больным с синдромом Гольденхара проводят УФО, ультразвук, магнитотерапию, лазерное лечение, оксигенотерапию.
В терапевтический комплекс входит также лечебная гимнастика, занятия с сурдологом и психологом, слухопротезирование цифровыми имплантируемыми слуховыми аппаратами, контроль слуха и периодические настройки аппаратов.
При сколиозе доктора назначают массаж и ношение специальных корсетов.
Лечение у ортодонта проходит в несколько этапов. Первый этап — молочный. Он представляет собой знакомство с патологией.
Специалисты объясняют родителям, как правильно ухаживать за полостью рта больного ребенка, предупреждают о возможных осложнениях, проводят аппаратное лечение для исправления дефектов челюсти.
Следующий этап — сменный, заключающийся в исправлении прикуса и коррекции имеющихся деформаций во рту. Постоянный этап – замена съемных аппаратов на брекеты и различные фиксаторы. Обычно к 18-летнему возрасту ортодонтическое лечение полностью завершается.
Народная медицина
Наиболее распространенные рецепты народной медицины:
- для промывания и очищения глаз можно использовать отвары и настои лекарственных трав – василька, подорожника, ромашки, тмина,
- употреблять внутрь средство, приготовленное из меда и сока калины,
- закапывать в уши настой из масла шиповника и семян аниса,
- принимать внутрь настой из корня аира.
Профилактика
Специфической профилактики синдрома Гольденхара не существует. К общим профилактическим мерам, позволяющим предотвратить это врожденное заболевание, относятся:
- отсутствие хронических гинекологических заболеваний у матери,
- правильное питание будущей матери,
- отказ родителей от вредных привычек,
- оптимальная физическая активность,
- обоюдное желание иметь ребенка,
- взаимопонимание, теплая и любящая атмосфера в семье.
Пренатальная диагностика врожденных аномалий заключается в проведении фетоскопии и ультразвукового сканирования эмбриона. Проводят УЗИ на 20-24 неделе беременности. Этот метод является точным на 100%. Он позволяет специалистам понять, стоит ли сохранять беременность.
Источник: https://sindrom.info/goldenxara/
Опасные последствия синдрома Гольденхара и методы лечения патологии

Синдром Гольденхара, или окуло-аурикуло-вертебральная дисплазия, – редкий порок развития наследственного характера. При заболевании поражаются кости скелета, нервно-мышечные компоненты и мягкие ткани, являющиеся производными 1 и 2 висцера́льных (жаберных) дуг, ответственных за формирование височно-нижнечелюстного сустава, челюстных и подъязычной костей, слухового аппарата.
В результате у больных наблюдается односторонняя гипоплазия лица, пороки в расположении и строении ушных раковин, нарушение слуха, дермоидные кисты, липомы глаз, деформация позвоночника.
Факторы, вызывающие патологию
Причины синдрома Гольденхара недостаточно изучены, считается, что виновником возникновения патологии в период внутриутробного развития становятся наследственные нарушения в гене, расположенном на длинном плече 14 хромосомы (в локусе 14q32), также отмечаются и другие хромосомные дефекты.
Предположительно, формирование диспластических повреждений окуло-аурикуло-вертебральной зоны возникает при возможном сосудистом инсульте у плода в районе 1 и 2 жаберных дуг при разрыве стременной артерии, когда происходит замена источника кровоснабжения. Кровоизлияние становится причиной нарушения процесса пролиферации клеток, что ведет к патологическим изменениям в формировании костей, мышц и иных составляющих мягких тканей.
Проявления патологии носят спорадический (единичный) характер, к факторам, повышающим вероятность рождения ребенка с синдромом Гольденхара, относят:
- кровосмесительные браки;
- тератогенное воздействие на ранних сроках вынашивания;
- диабет матери;
- избыток веса женщины.
Показатель риска появления больного ребенка у носителя аномального гена равняется 3%, повторные случаи рождения в семье отмечаются с частотой в 1%.
Синдром диагностируется у 1 новорожденного из 3,5–5,6 тысяч живых младенцев, выявляется у 1 ребенка из 1 тыс. глухих детей. У мальчиков встречается на треть чаще, чем у девочек.
Проявления патологии
Односторонние нарушения в районе 1 и 2 хрящевых пластин при развитии эмбриона приводит к диспропорции в формировании черепно-лицевого комплекса .
Клиническая картина при синдроме Гольденхара варьируется от незначительной асимметрии лица вплоть до тяжелого недоразвития челюстей, височной кости, глазной орбиты и ушной раковины. Патология проявляется следующими симптомами:
- Дефекты развития уха. Отмечаются в 80% случаев, характеризуются деформацией или дисплазией ушной раковины, иногда ее отсутствием (анотия). У 50% больных наблюдается аномальное расположение органа слуха, также могут появляться околоушные свищи. Из-за поражения среднего уха и слухового нерва тугоухость или глухота диагностируется у 30–50% детей.
- Офтальмологические мальформации включают образование на глазном яблоке опухолевых элементов: на конъюнктиве – дермоидов и липодермоидов, на верхнем веке – колобомы. Иногда выявляется страбизм, катаракта, повреждения глазодвигательной мускулатуры, глаз может быть недоразвит или полностью отсутствовать.
- Челюстные пороки. Гипоплазия челюстей, недоразвитие отростков нижней челюсти присущи 85% с синдромом Гольденхара, также наблюдается недостаточное формирование мышц лица, губ. Встречается высокая арка нёба, в некоторых случаях с расщелиной, открытый прикус, добавочные уздечки, трахеопищеводный свищ. Широкая щель рта, при этом один угол приподнят.
Синдром сопровождают и другие расстройства: почти у половины больных наблюдаются повреждения в развитии позвонков, преимущественно шейного отдела, у трети детей выявлялись дефекты ребер, у 20% отмечается косолапость. У трети маленьких пациентов обнаруживаются повреждения в работе сердечно-сосудистой системы. Встречаются случаи с патологиями органов: легких, почек и матки.
У 10% детей выявляются нарушения в деятельности ЦНС и умственная отсталость.
Диагностические мероприятия
Типичная асимметрия лица в совокупности с видимой симптоматикой обычно не вызывает затруднений в выявлении синдрома Гольденхара, поэтому предварительный диагноз выставляется сразу после рождения на этапе визуального осмотра. Для подтверждения патологии назначаются следующие виды обследования:
- Так как поражение среднего и наружного уха относятся к типичным проявлениям болезни, у детей проверяется острота слуха на обоих ушах. Используется метод компьютерной аудиометрии, а также применяется электрокохлеография, импедансометрия. У детей постарше аудиометрию проводят в игровой форме. До 7 лет обследование слуха необходимо делать регулярно каждые полгода.
- Для выявления деформации черепа и позвоночника назначается рентгенография.
- Нарушения во внутренних органах определяют при помощи УЗИ.
- Изменения в работе сердца диагностируют на основе электрокардиограммы.
- КТ височной области назначается детям после 3-х лет.
Для консультации приглашаются узкие специалисты: генетик, сурдолог, дефектолог, офтальмолог, логопед, нефролог, невролог, челюстно-лицевой хирург.
Современные методы лечения
Вариативность нарушений, разнообразие клинической картины и различная степень выраженности дефектов развития требуют индивидуального многоэтапного и мультидисциплинарного подхода в лечении синдрома Гольденхара. Вид мероприятий зависит от степени повреждений и возраста больного.
В легких случаях патологии до 2-х или 3-х лет осуществляется наблюдение за ребенком, далее по показаниям применяется ортодонтическая терапия. Метод лечения – подготовка зубного ряда, лицевых и жевательных мышц к оперативному вмешательству, меры направлены на исправление дефектов прикуса и предупреждение прогресса асимметрии лица, осуществляются в 3 этапа:
- Молочный прикус. Родителей знакомят с особенностями болезни, учат ухаживать за полостью рта ребенка. Период предусматривает ношение съемных одночелюстных или двучелюстных аппаратов, изготовленных из эластичного материала по индивидуальному заказу.
- Сменный прикус. Для устранения нарушений прикуса используются съемные удерживающие и расширяющие устройства, а также несъемные каркасные конструкции.
- Сменный поздний и постоянный прикус. Устанавливаются брекет-системы для длительного ношения.
В ретенционный период, длящийся до окончания роста ребенка, закрепляется достигнутый результат, больному ставятся металлические ретейнеры, на ночь фиксируется ретенционный аппарат.
Возможные осложнения
Последствия болезни зависят от степени выраженности дефектов развития. Тяжелые формы нарушений в некоторых случаях несовместимы с жизнью и приводят к гибели младенца после рождения. Если заболевание сопровождается не только внешними проявлениями, но и изменениями во внутренних органах, умственной отсталостью, ребенку с синдромом Гольденхара дают инвалидность.
Своевременная диагностика, комплексное лечение и полноценная реабилитация в 75% случаев помогают избежать формирования осложнений. Впоследствии больные общаются, работают и учатся, создают семьи. Если время упущено, прогресс гипоплазии костей лица усугубляется:
- ребенку становится сложнее жевать и совершать глотательные движения;
- прогрессируют расстройства слуха и зрения;
- в результате нарушений формируется ряд физических неудобств.
В итоге дети и родители испытывают психологический дискомфорт и сложности с адаптацией в социуме.
Гольденхара синдром: описание, причины, симптомы, наследование и лечение

Гольденхара синдром – это наследственное заболевание окулоаурикуловертебральной области, то есть поражение структур, исходящих из первой и второй жаберных дуг.
Оно было описано в 1952 году американским врачом Гольденхаром.
Это наследственное заболевание человека, при котором чаще всего ярко выражена односторонняя гипоплазия лица, аномальные ушные раковины, веки, зубы, позвоночник и т. д.
Синдром Гольденхара
Синдром Гольденхара был описан впервые в середине двадцатого века. Но выделен в отдельное заболевание несколько позже американским доктором, по фамилии которого и получил свое название. Частота развития составляет 1:3000-5000. По всему миру фиксируется мало случаев подобного заболевания.
Причины появления болезни
Гольденхара синдром изучен еще недостаточно. Но большинство ученых сходятся во мнении, что причиной его появления являются гены. В большинстве случаев характер заболевания спорадический.
Почему еще может возникнуть синдром Гольденхара? Причины могут крыться и в кровном родстве родителей. Но большую роль при возникновении синдрома могут сыграть прежние аборты матери больного ребенка или тератогенные факторы, которые имели место на ранних сроках беременности. Помимо этого причиной рождения ребенка с таким синдромом вполне может быть ожирение или сахарный диабет у матери.
Наследование синдрома Гольденхара передается по аутосомно-рецессивному или доминантному типу. Происходят перестройки в некоторых хромосомах. Иногда при появлении синдрома выявляются мутации гена GSC. Если мутация затронет еще и ген TCOF1, то это приводит к развитию дополнительного синдрома Тричера-Коллинза.
Диагностика синдрома
Для диагностики синдрома Гольденхара применяется множество процедур. Одна из них – исследование слуха. Это помогает определить степень глухоты на пораженной стороне. Сделать это необходимо как можно быстрее, чтобы ребенок впоследствии не отставал в психоречевом развитии.
В раннем возрасте у детей проверяются слуховые потенциалы, ответ мозга и модулированные тоны. Обследуют ребенка во время сна. К старшим детям для диагностики слуха применяется речевая аудиометрия во время игры. При таком синдроме обычно выявляется тугоухость разной степени. Диагностика слуха должна проводиться раз в полгода в течение семи лет.
Кроме перечисленного используется компьютерная томография височных зон, которая проводится для детей старше трехлетнего возраста. Ребенок, имеющий синдром Гольденхара, описание патологий которого представлено несколько выше, должен проходить постоянное обследование у врачей:
- кардиолога;
- логопеда;
- генетика;
- хирурга;
- ортопеда;
- дефектолога;
- офтальмолога;
- невропатолога.
Гольденхара синдром: ортодонтическое лечение
Ортодонтическое лечение разделяется на три этапа прикуса:
- Молочного.
- Сменного.
- Постоянного.
Первый этап самый важный. Во время него родителям впервые сообщается о пороке, степени его тяжести и возможных последующих рисках и развитии болезни. Сообщаются обязательные правила гигиены ротовой полости, которые ребенок должен соблюдать неукоснительно.
Если малыш не получил своевременного лечения сразу, то с возрастом у него может проявиться недоразвитие костей лица. Причем оно будет прогрессировать. Аномальный прикус со временем станет все более заметным. Глотать и жевать ребенку будет все труднее.
И как результат перечисленных ухудшений – психологическая травма родителей и ребенка.При лечении на первом этапе применяются съемные эластичные челюстные и пластинчатые аппараты. Это время, когда главная цель врачей – познакомить ребенка со специальными аппаратами, помочь ему адаптироваться и привыкнуть к их использованию. Если асимметрия изначально не выражена, то такие приборы имеют предупреждающий ее характер.
Второй этап лечения
Главные задачи – устранить аномалии прикуса. Исправить неправильные формы и размеры зубоальвеолярных дуг. Продолжается работа по предупреждению и устранению порока челюсти и ее недоразвития. Для лечения применяются съемные эластичные пластины. Эти аппараты служат опорой и поддержкой для челюстей. Дополнительно используются несъемные каркасные.
Третий этап
На третьем этапе продолжается все перечисленное выше лечение. Но начинают использовать уже несъемные брекеты. В конце лечения применяется ретенционный период, во время которого проходит закрепление достигнутых прежде результатов. Используются несъемные ретейнеры из проволоки, фиксирующие верхнее нёбо и зубы. Для окружающих такие аппараты уже незаметны.
Одновременно ребенок получает съемный фиксатор, который используется только во время сна. Ретенционный период заканчивается, только когда ребенок перестает расти.
За это время проводятся периодические обследования. Ребенок находится под неусыпным контролем врачей. Если необходимо, делаются пластические операции.
Реабилитация ребенка заканчивается только к 18 годам, то есть когда его тело уже полностью сформировано.
Источник: https://FB.ru/article/218986/goldenhara-sindrom-opisanie-prichinyi-simptomyi-nasledovanie-i-lechenie
Врожденные деформации

Cинонимы: синдром первой и второй жаберных дуг, синдром Гольденхара – окуло-ауриколо-вертебральная дисплазия, краниофациальная микросомия, отомандибулярный дизостоз и латеральная фациальная дисплазия — является редким наследственным заболеванием, характеризующимся значительным числом аномалий, которые возникают вследствие нарушения развития первой и второй жаберных дуг первого глоточного кармана, первой жаберной щели и зачатков височной кости. Среди врожденных пороков развития черепно-челюстно-лицевой области гемифациальная микросомия занимает второе место по частоте встречаемости после врожденных расщелин верхней губы и нёба. Частота этого синдрома колеблется в пределах 1:3500-5600 новорожденных, он присутствует у 1 из 1000 у детей с врожденной глухотой. Распределение по половому признаку (мужчины и женщины) составляет примерно 3:2.
Этиология и тип наследования изучены недостаточно.
Неблагоприятный акушерско-гинекологический анамнез матери (предшествующие аборты, сахарный диабет, избыточный вес) и тератогенные факторы на ранних сроках беременности являются отягощающими факторами риска рождения больного ребенка. Вероятность повторного рождения больного ребенка, как и вероятность рождения больного ребенка у носителя патологии, ориентировочно равна 2%.
Клинически для гемифациальной микросомии характерны: недоразвитие тела и ветви нижней челюсти, гипоплазия скуловой кости и дуги, недоразвитие структур ВНЧС; аплазия ветви нижней челюсти и ВНЧС; нарушение размеров и положения глазницы; гипоплазия, аплазия ушной раковины, атрезия слухового прохода, поражение лицевого нерва, гипоплазия мимических мышц; дефицит мягких тканей; макростомия; предушные придатки и свищи; иногда сочетание с врожденной расщелиной губы и неба, эпибульбарным дермоидом, аномалией прикуса, адентией, нарушением структуры эмали и формы зубов, пороками развития опорно-двигательного аппарата, органов зрения и нервной системы, а также аномалиями мочевыделительной системы и желудочно-кишечного тракта.
Cклеродермия
Это прогрессирующее системное заболевание, в основе которого лежит воспалительное поражение мелких сосудов всего организма, с последующими фиброзно-склеротическими изменениями кожи, опорно-двигательного аппарата и внутренних органов.
При очаговой склеродермии наблюдается ограниченное уплотнение кожи, но могут вовлекаться подкожные ткани и кости. Выделяют две основные формы очаговой склеродермии – бляшечную (морфея) и линейную.
В первом случае поражение кожи имеет вид уплотнений округлой формы («Бляшки»), с лиловым ободком по периферии в дебюте болезни. Эти очаги, единичные или множественные, могут появляться как на туловище, так и на лице и конечностях.
При линейной форме очаговой склеродермии участки поражения имеют вид полос уплотнения кожи, часто с вовлечением подлежащих мышц и костей, и локализуются, главным образом, на конечностях и лице.
Эта форма очаговой склеродермии в случае развития в детском и подростковом возрасте, может приводить к ограничению движений (мышечные и суставные контрактуры) и нарушениям развития пораженных участков. Внутренние органы при очаговой склеродермии не страдают.Системная склеродермия (ССД) – форма склеродермии, при которой помимо уплотнений кожи развиваются разнообразные поражения суставов, внутренних органов (сердца, легких, желудочно-кишечного тракта, почек).
В редких случаях наблюдается поражение только внутренних органов, без изменений кожи. Женщины заболевают в 3-5 раза чаще, чем мужчины.
ССД подразделяется на лимитированную и диффузную форму, которые различаются по распространенности и выраженности поражения кожи и внутренних органов.
Липодистрофия
Общее или локальное поражение подкожной клетчатки с уменьшением (атрофическая форма) или увеличением (гипертрофическая форма) объема жировой ткани. Л. могут быть генерализованными или сегментарными.
К липодистрофиям относят следующие патологические состояния: врожденную генерализованную Л., гипермускулярную Л., прогрессирующую сегментарную Л., или болезнь Барракера — Симонса, постинъекционную Л.
, Липоматоз болезненный (болезнь Деркума).
Прогрессирующая сегментарная липодистрофия (синонимы липоатрофия, болезнь Барракера-Симона) — это Относительно редкое заболевание, проявляющееся атрофией подкожной жировой клетчатки лица, шеи, плечевого пояса, грудной клетки при нормальном или избыточном отложении жира в нижней половине тела.
Встречается редко, поражает преимущественно женщин. Этиология неясна. Внутренние органы не поражаются, функция их не нарушена. Больные обращаются к врачу из-за своей внешности, иногда жалуются на слабость, раздражительность. Жизненный прогноз благоприятен.
Специфичного лечения нет, применяют средства, укрепляющие нервную систему, и витамины.
Микрогения
Это аномалия развития лица, характеризующееся гипоплазией (недоразвитием) нижней челюсти.
В основе деформации лежит нарушение роста и развития нижней челюсти в результате травмы или патологического процесса в области сустава и ветви челюсти, перенесенных во время родов, в детском возрасте при незаконченном росте лицевого скелета, а также вследствие врожденных нарушений во время развития нижне- и верхнечелюстного отростков головной части зародыша. При односторонней микрогении лицо асимметрично за счет укорочения ветви и горизонтальной части тела челюсти, что приводит к смещению подбородка в сторону укорочения. Пораженная сторона выглядит более выпуклой, здоровая сторона уплощена, тело челюсти удлинено и деформировано. Двусторонняя (симметричная) микрогения характеризуется укорочением тела и ветвей челюсти, что сопровождается смещением подбородочного отдела кзади и его «скошенностью». При этом верхняя челюсть выступает вперед, придавая лицу так называемое «птичье выражение». Деформация нижней челюсти может быть выражена в различной степени: от незначительной асимметрии или уплощения подбородочного отдела с умеренным нарушением прикуса до тяжелых, обезображивающих деформаций. При выраженном нарушении прикуса развиваются функциональные расстройства из-за затруднения акта откусывания и пережевывания пищи.
Микрогения довольно часто сочетается с анкилозом височно-челюстного сустава.
Источник: https://facesurgery.pro/face-surgery/xirurgiya-deformaczij/vrozhdennyie-deformaczii.html