
Синдром клайнфельтера
Содержание
Синдром Клайнфельтера: основные причины, первые симптомы, лечение и прогноз
Синдром Клайнфельтера — наследственное заболевание. Клиническая картина синдрома описана в 1942 году в работах Гарри Клайнфельтера и Фуллера Олбрайта.
Синдром Клайнфельтера — генетический недуг, который диагностируют исключительно у мужчин. Его характерный признак – наличие добавочной женской половой хромосомы в мужском кариотипе ХY. Проявляется недуг развитием различных эндокринных нарушений. У человека с таким диагнозом наблюдается неполноценная и недостаточная выработка мужских половых гормонов.
Синдром Клайнфельтера не является редким болезненным состоянием. Его диагностируют в год у 1 из 800 новорождённых мальчиков. Часто провести полноценную диагностику недуга не удаётся, что в дальнейшем становится причиной тяжёлого его протекания. Обычно болезнь приводит к бесплодию, поэтому можно сказать, что из поколения в поколение она не передаётся.
Распространенность заболевания
Синдром Клайнфельтера является одним из наиболее распространенных генетических заболеваний. Около 0,2% мужского населения Земли страдает этой патологией. Кроме того, синдром Клайнфельтера – третья по распространенности эндокринная патология у мужчин (после сахарного диабета и гиперфункции щитовидной железы).
На сегодняшний день синдром Клайнфельтера является наиболее частой причиной врожденного нарушения репродуктивной функции у мужчин. По статистическим данным, около половины случаев синдрома Клайнфельтера остаются нераспознанными. Нередко такие пациенты обращаются за помощью по поводу различных нарушений (бесплодие, нарушения эректильной функции, гинекомастия, остеопороз и др.
), однако основное заболевание остается недиагностированным.
Этиология
Синдром Клайнфельтера обусловлен мутацией генов, приводящей к удвоению женской половой хромосомы в кариотипе мужчины. Нерасхождение половых хромосом в процессе мейоза либо митоза может быть обусловлено различными факторами. Самыми распространенными среди них являются:
- Вирусы,
- Неполноценность иммунной системы матери или отца,
- Плохое экологическое состояние окружающей среды,
- Дети от родственных браков,
- Ранний или поздний возраст матери,
- Наследственные патологии в предыдущих поколениях.
Мальчики с синдром Клайнфельтера вместо нормального мужского генотипа ХУ приобретают одну У хромосому и несколько Х хромосом. Такое изменение генетического набора приводит к появлению особых внешних данных, незначительному снижению интеллекта и развитию целого ряда сопутствующих заболеваний.
Повышение концентрации фолликулостимулирующего и лютеинизирующего гормонов в крови приводит к фиброзу, гиалинозу и атрофии семенных канальцев. Яички перестают развиваться, становятся маленькими и плотными. Облитерация семенных канальцев заканчивается развитием азооспермии и бесплодия.
Признаки и симптоматика
В отличие от большего числа различных болезней с нарушением хромосомного набора, внутриутробное развитие малышей с таким диагнозом проходит вполне нормально. Это говорит о том, что ни в младенческом, ни в раннем детском возрасте, заподозрить прогрессирование генетического недуга невозможно. Первые его признаки могут проявиться в препубертатном периоде:
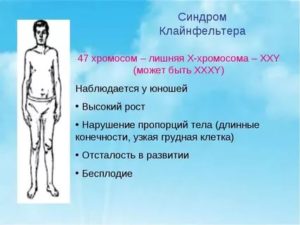
- мальчики от 5 до 8 лет отличаются высоким ростом (относительно своих сверстников);
- талия высокая;
- ноги и руки удлинённые;
- нарушение восприятия информации на слух.
Признаки недуга в подростковом периоде:
- гинекомастия — увеличение груди;
- на лице отмечается незначительная растительность или она может полностью отсутствовать;
- волосы в лобковой зоне отрастают по женскому типу (треугольником);
- оволосение на груди и прочих частях тела отсутствует;
- маленькие яички.
По достижению 25 лет, больные начинают предъявлять жалобы на:
- снижение либидо и потенции;
- мужское бесплодие;
- остеопороз;
- ожирение;
- возможно также развитие сахарного диабета, ревматоидного артрита.
Симптомы болезни напрямую зависят от сформированного кариотипа. Степень их выраженности зависит от того, сколько дополнительных хромосом имеет этот самый кариотип. Симптомы кариотипа 48 ХХYY:
- высокий рост. В большинстве случае рост пациента выше 182 см;
- агрессивность;
- снижение интеллекта;
- дисфории;
- склонность к депрессии;
- замедленная речь.
Симптомы недуга с кариотипом 48 ХХХY:
- рост средний или высокий;
- глазной гипертелоризм;
- плоская переносица;
- клинодактилия 5 пальца;
- снижение интеллекта;
- апатия;
- агрессия отсутствует.
Симптомы патологии в случае кариотипа 49ХХХХ:
- переносица становится практически плоской;
- глазной гипертелоризм;
- микроцефалия (голова маленькая по сравнению с телом);
- глазная щель узкая;
- аномалии развития верхних и нижних конечностей (искривление формы стоп, коленных суставных сочленений);
- вспышки гнева;
- агрессивность.
Симптомы мозаичной формы недуга такие же, как и при классической. Единственное, что стоит отметить отдельно, это то, что при этой форме у пациента сохраняется возможность иметь детей. Конечно, она снижена, но все же присутствует.
Лечение
При клинически выраженном синдроме Клайнфельтера необходима пожизненная заместительная терапия препаратами мужского гормона тестостерона. Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни.
Кроме того, заместительная терапия предупреждает остеопороз и мышечную слабость, а в случае их развития способна вернуть кости и мышцы пациента в нормальное состояние. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза, поскольку это необходимо для профилактики гинекомастии.
Дело в том, что уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения. Нередко пациенты испытывают сильный психологический дискомфорт из-за увеличенных грудных желез, так что приходится прибегать к хирургической коррекции (мастэктомия).Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Профилактика синдрома и прогнозирование
Пациенты, страдающие синдромом Клайнфельтера, имеют такую же продолжительность жизни, как и другие люди, правда, наличие склонности к появлению хронических заболеваний может оказаться фактором наступления ранней смертности. Большинство таких больных оказываются бесплодными.
Единственно вероятным вариантом рождения детей в семье, в которой партнер болен, служит использование донорской семенной жидкости.
Но, тем не менее, при наличии мозаичной формы синдрома Клайнфельтера мужчина все же может стать отцом самостоятельно либо, использовать вспомогательную репродуктивную технологию, к примеру, экстракорпоральное оплодотворение.
Для оценивания вероятности рождения ребенка с этим синдромом в процессе наблюдения беременности женщинам предлагают проходить пренатальный скрининг. Правда, даже в случаях получения положительных сведений о наличии такого синдрома у плода, настаивание на том, чтобы женщина прервала беременность, со стороны гинеколога является абсолютно недопустимым.
Таким образом, решение вопроса о целесообразности продолжения беременности должны принимать исключительно будущие родители. При наличии нормального кариотипа у родителей риск появления ребенка с подобной хромосомной аномалией составляет не больше одного процента.
Диспансерное наблюдение пациентов, страдающих синдромом Клайнфельтера, осуществляют эндокринологи.
(1 5,00 из 5)
Загрузка…
Источник: https://osindromah.ru/geneticheskie/sindrom-klajnfeltera.html
Синдром Клайнфельтера у детей и подростков
Синдром Клайнфельтера (СК) — врожденное генетическое заболевание лиц мужского пола, обусловленное наличием в мужском кариотипе дополнительной половой Х-хромосомы (одной или нескольких).
Данный синдром характеризуется многообразием цитогенетических вариантов: 47, XXY, 48, XXXY, 48, XXYY, 49, XXXXY, 49, XXXYY, 46, XY/47,XXY.
«Классическим» кариотипом при СК является 47, XXY, который встречается примерно в 90% случаев; мозаичные формы составляют 7%, остальные варианты полисомий — около 3% [1].
Распространенность СК крайне высока: колеблясь от 1:500 до 1:1000 (0,1—0,2%) новорожденных мальчиков [1, 2]. В 3% случаев СК выявляется у мужчин с бесплодием, в 11% — при азооспермии.
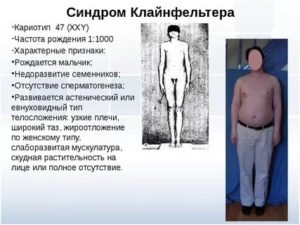
Присутствие в кариотипе дополнительной Х-хромосомы приводит к клинико-лабораторным проявлениям, из которых самыу частые бесплодие (91—99%) и азооспермия (>95%), малый (95%), высокий уровень гонадотропинов (>95%) и низкий уровень тестостерона в крови (63—85%), скудное оволосение лица и тела (60—80%), трудности обучения (>75%), легастения (затруднение приобретения навыков чтения и письма 50—70%), гинекомастия (38—75%), высокорослость (30%) [3].
В 1942 г. Harry Fitch Klinefelter (в честь которого будет назван синдром), Edward C.
Reifenstein и Fuller Albright [4] обнаружили у 9 мужчин в возрасте от 17 до 38 лет ранее не описанное сочетание: двусторонней гинекомастии, маленького размера яичек (при нормальном развитии полового члена) и азооспермии с повышенной экскрецией фолликулостимулирующего гормона (ФСГ) с мочой. Вначале авторы предполагали, что гинекомастия у пациентов является следствием повышенной секреции эстрогенов, но в ходе обследования эта гипотеза была опровергнута. Активно обсуждалось наличие гипотетического Х-гормона (ингибина B), секретируемого тестикулами, и высказывалось предположение о дефиците этого гормона в качестве причины гиперсекреции ФСГ и гинекомастии. Механизм формирования азооспермии, гиалинизации семенных канальцев оставался неясным.
В 1949 г. M. Barr и E. Bertram [5] при изучении ядер кошачьих нейронов обнаружили наличие в клетках самок и отсутствие у самцов маленького тельца, расположенного близко к ядру (позже получившего название «половой хроматин», или «тельце Барра»). В 1955 г. K. Moore и M. Barr [6] описали простой метод изучения полового хроматина — в мазке со слизистой оболочки щеки. J.
Bradbury и соавт. [7] в 1956 г.
при исследовании буккального эпителия 19-летнего пациента с признаками СК (малый объем тестикул, нормальное развитие полового члена, лобковое оволосение по женскому типу, двусторонняя гинекомастия, высокий уровень гонадотропинов в моче, отсутствие сперматозоидов в эякуляте и склероз семенных канальцев по данным биопсии) обнаружили наличие полового хроматина.
В 1959 г. P. Jacobs и J. Strong [8] выполнили стернальную пункцию у 24-летнего мужчины с гинекомастией и гипоплазией тестикул и в 44 изученных клетках обнаружили по 47 хромосом.
В связи с тем что у данного пациента был выявлен также половой хроматин, а размер дополнительной хромосомы соответствовал размеру хромосомы Х, авторы предположили, что при СК в кариотипе имеется дополнительная Х-хромосома, что и подтвердилось впоследствии.
Количественное нарушение хромосомного набора, приводящее к СК, является следствием нерасхождения хромосом в процессе родительского гаметогенеза. Дополнительная Х-хромосома с равной вероятностью имеет отцовское и материнское происхождение [9].
В ходе сперматогенеза ошибка расхождения хромосом происходит только в процессе первого мейотического деления (при формировании сперматоцитов второго порядка), тогда как в ходе овогенеза ошибки могут происходить на разных уровнях клеточного деления [10]:
1) в процессе мейоза I (при формировании ооцитов второго порядка) — частота нерасхождения хромосом на данном этапе составляет примерно 48% в структуре материнского наследования;
2) в процессе мейоза II вследствие ошибки разделения сестринских хроматид при формировании яйцеклетки — 29%;3) в процессе постзиготического митотического деления —16%;
4) на этапе мейотического деления, стадия которого не определена — 7% (см. рисунок). Схема образования дополнительной Х-хромосомы в ходе гаметогенеза. 1 — на стадии мейоза I у матери; 2 — на стадии мейоза II у матери; 3 — в процессе постзиготического митотического деления; 4 — на стадии мейоза I у отца [34].
Среди факторов риска хромосомных нарушений в процессе деления клеток выделяют возраст матери старше 40 лет, особенно для этапа дробления зиготы [1]. Влияние возраста отца остается спорным [11].
Несмотря на высокую распространенность СК в популяции, заболевание только в 10% случаев диагностируется до начала пубертата и только в 25% — в течение жизни мужчины [1].
Причиной позднего выявления данного состояния являются выраженная вариабельность симптомов, различное время их появления и степень выраженности, в связи c чем пациенты могут долгое время наблюдаться у разных специалистов, не зная об основном диагнозе.
Клинико-лабораторные признаки СК в детском возрасте
У большинства детей с СК определяются антропометрические показатели, не отличающиеся от нормы [12, 13]. Среди признаков, позволяющих заподозрить СК у новорожденных, описывают неправильно развитые гениталии, крипторхизм (с частотой встречаемости 27—37%), маленький объем яичек и микропенис (частота 10—25%) [3, 13].
В течение первых 2 лет жизни могут иметь место задержка речевого развития (появление первых слов только в 18—24 мес) и более позднее начало самостоятельной ходьбы (≈18 мес) [14]. По данным С. Samango-Sprouse [15], у 68% детей с СК в возрасте от 2 мес до 7 лет выявляется мышечная гипотония.
Описаны также дискоординация движений, умеренно выраженная гипермобильность суставов, плоскостопие с пронацией голеностопного сустава и вальгусная деформация коленных суставов, искривление дистальной фаланги V пальца кисти [16].
Ускорение роста — один из характерных признаков СК — у большинства детей начинает обращать на себя внимание между 5-ю и 8-ю годами жизни [12]. У детей младшего школьного возраста в 40% выявляются нарушения речевого развития, включающие трудности выражения мыслей и проблемы вербальной коммуникации.Более 75% детей испытывают затруднения в приобретении навыков чтения и письма [3]. Следует отметить, что именно особенности психического развития и поведения у детей с СК чаще всего служат основанием для проведения кариотипирования в этом возрасте [17].
У многих пациентов с СК при кариотипе 47, ХХY значительные нарушения интеллектуального развития отсутствуют (IQ варьирует от 90 до 100 баллов) [10]. Однако с присутствием в кариотипе каждой следующей дополнительной Х-хромосомы выраженность нарушений речевого развития возрастает, а IQ снижается примерно на 15—16 баллов [12].
Среди особенностей психического развития у детей с СК выделяют эмоциональную лабильность, высокую тревожность, склонность к депрессивным состояниям, низкую мотивацию к деятельности и заниженную самооценку [18].
H. Bruining и соавт. [19] при обследовании 51 ребенка с СК в возрасте от 6 до 19 лет в 27% случаев выявили расстройства аутистического спектра, что еще больше может затруднять приобретение социальных навыков.
У большинства мальчиков с СК в допубертатном возрасте определяются нормальные уровни тестостерона, ЛГ, ФСГ, ингибина В, АМГ, СССГ.
Повышенный уровень ФСГ и сниженный уровень ингибина В (отражающий нарушение функции клеток Сертоли) регистрируются только у 10—20% больных [20].
Ряд авторов [14, 21] выявили снижение пиковой секреции тестостерона у детей с СК в период мини-пубертата, что может свидетельствовать о ранней манифестации тестикулярной недостаточности у отдельных пациентов с С.К.Сообщается и о высоконормальных уровнях тестостерона у мальчиков с СК в возрасте 3 мес [22]. В этой же работе продемонстрировано повышение соотношения ФСГ/ингибин В (6,5 у детей с СК против 3,0 у детей контрольной группы). Предполагается, что увеличение данного показателя отражает раннюю дисфункцию клеток Сертоли.
Таким образом, у подавляющего большинства детей с СК до наступления пубертата определяются нормальные показатели гормонального профиля, хотя у части пациентов уже в раннем возрасте могут быть обнаружены признаки тестикулярной недостаточности.
Клинико-лабораторные признаки СК в периоде пубертата
Среди клинических признаков СК в подростковом возрасте могут отчетливо проявляться диспропорции тела (длина ног начинает превышать длину туловища), снижение мышечной массы, скудное оволосение лица и груди, маленький объем тестикул, гинекомастия.
Рост увеличивается преимущественно за счет длины ног, которая при СК в среднем на 5,7 см превышает аналогичный показатель у детей контрольной группы [23].
Пропорции тела при СК несколько отличаются от характерных для других видов гипогонадизма, поскольку размах рук в большинстве случаев не превышает длины тела [24].
Увеличение грудных желез определяется примерно у трети взрослых мужчин с СК [23]; оно начиная обращать на себя внимание, как правило, в пубертатном периоде. Одним из предполагаемых механизмов развития гинекомастии считается повышенное отношение эстрадиола к тестостерону [25].
L. Aksglaede и соавт. [26] выявили гинекомастию у 16 (47%) из 34 подростков с СК, а S. Close и cоавт. [18] — у 5 (18,5%) из 27, тогда как ни у одного из 16 детей допубертатного возраста гинекомастия не отмечалась. Сходные результаты были получены N. Pacenza и соавт. [27]: гинекомастия была выявлена у 11 (42,3%) из 26 подростков с СК и не у одного из 18 детей младшего возраста.
В отличие от физиологической юношеской гинекомастии, часто развивающейся в период полового созревания и имеющей транзиторный характер, гинекомастия у подростков с СК, как правило, постоянна.
Эффективность медикаментозного лечения гинекомастии (например, ингибиторами ароматазы) при СК остается недоказанной [16]. Поэтому при выраженном дискомфорте из-за увеличенного размера грудных желез следует рассматривать возможность хирургического вмешательства.
Несмотря на повышенный риск рака грудных желез при СК (частота встречаемости 0,3%), гинекомастию не считают предрасполагающим к раку фактором [24].Возраст начала пубертата и раннее его течение у большинства мальчиков с СК существенно не отличаются от нормы: в период с 11 до 14 лет объем тестикул увеличивается примерно до 6—8 мл, уровень тестостерона в крови превышает 10 нмоль/л, обеспечивая увеличение длины полового члена и лобковое оволосение. Половое развитие по Таннеру достигает IV—V стадии [27—29]. Однако в дальнейшем объем тестикул уменьшается (до 1,7 ммоль/л), снижение уровня липопротеинов высокой плотности (ЛПВП) до
Источник: https://www.mediasphera.ru/issues/problemy-endokrinologii/2018/5/1037596602018051321
Синдром Клайнфельтера: причины, признаки, диагностика, как лечить
Синдром Клайнфельтера — распространенный генетический недуг, обусловленный дефицитом гормона тестостерона. Клиническую картину и особенности патологии впервые описали в 1942 году американские врачи Г. Клайнфельтер и Ф. Олбрайт.
В честь одного из них аномалия получила свое название. Эта хромосомная болезнь регистрируется у одного или двух мальчиков из тысячи новорожденных. У больных в кариотипе присутствует хотя бы одна дополнительная женская хромосома.
Патология не создает проблем таким детям до начала пубертатного периода.
Синдром Клайнфельтера — самая распространенная генетическая патология, а также не менее популярное эндокринное заболевание, которое часто остается нераспознанным. В структуре эндокринной патологии оно уступает лишь сахарному диабету и тиреотоксикозу.
Первичная форма гипогонадизма сопровождается плохим функционированием секреторной ткани яичек, неполным развитием внутренних и наружных половых органов, недоразвитием вторичных половых признаков, нарушением белкового и жирового обмена веществ.
Заболевание не передается по наследству, поскольку больные мужчины абсолютно бесплодны.
Диагностика синдрома Клайнфельтера основывается на данных кариотипирования, результатах определения в крови половых гормонов, спермограммы, УЗИ мошонки и морфологического изучения биоптата яичек. В настоящее время этиологическое лечение хромосомных аномалий не разработано.
Современные терапевтические методики позволяют сгладить основные симптомы болезни и нормализовать социальную жизнь носителей более двух половых хромосом. Больным назначают гормоны, проводят оперативное вмешательство — мастэктомию. Синдром Клайнфельтера – неизлечимая патология.
Клиника
Мальчики с синдромом Клайнфельтера при рождении имеют нормальный рост и вес. Клинические признаки заболевания у них тоже отсутствуют: мошонка и половой член располагаются правильно и симметрично. Обнаружить патологию у новорожденных и грудных детей невозможно. Клиника синдрома Клайнфельтера становится явной только после начала пубертатного периода.
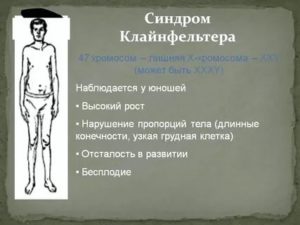
Заподозрить заболевание можно и раньше по ряду характерных признаков. У больных мальчиков ноги длиннее, чем у сверстников, талия расположена несколько выше, мускулатура развита слабо. Узкие плечи и широкие бедра придают фигуре определенную женственность. Отложение жира также происходит по женскому типу.
Диспропорциональное телосложение — один из характерных признаков данного наследственного заболевания.
Мальчики с синдромом Клайнфельтера подвержены часто возникающим вирусным респираторным заболеваниям. Это первый тревожный признак, заставляющий родителей и педиатров обратить особое внимание на ребенка. Больные дети поздно начинают ползать, сидеть, ходить, говорить.
Мальчики заметно прибавляют в росте между 6—7 годами. Большинство из них имеют атипичное строение лица. У больных детей часто заметно снижен интеллект и нарушено психическое развитие. Они не успевают в школе, быстро утомляются, плохо воспринимают устную речь, редко заводят друзей, избегают общения с незнакомыми людьми.
Психика пациентов лабильна: часто возникают периоды полного равнодушия к происходящим событиям, чередуются радость и печаль, настроение меняется по малейшему поводу. Мальчики плохо адаптируется в социуме.
Одни становятся скромными, замкнутыми, тихими и апатичными, другие — импульсивными и резкими, у третьих появляются склонности к криминалу.
У подростков развивается андрогенный дефицит, который проявляется определенными симптомами:
- У больных увеличиваются грудные железы. Развивается двусторонняя, безболезненная и необратимая гинекомастия, сохраняющаяся пожизненно. Ранняя гормонотерапия снижает выраженность гинекомастии. Половые гормоны необходимо принимать сразу после постановки диагноза.
- У большинства детей обнаруживают патогномоничный признак заболевания – маленькие и плотные яички. При этом половой член имеет размеры, не соответствующие возрасту. Сформированная мошонка часто без складок и пигментации. Простата не обнаруживается при пальпации. Гипогонадизм и гипогенитализм развиваются у всех больных.
- Волосы отсутствуют на лице и груди, растут на лобке треугольником. Вторичные половые признаки появляются очень поздно. Ухудшаются показатели спермограммы. Недоразвитие гортани проявляется высоким голосом у больных мужчин.
- Крипторхизм или неопущение яичек – врожденная аномалия, которая часто имеется у детей с синдромом Клайнфельтера. Мошонка становится асимметричной, в ней отсутствуют яички, возникает ноющая боль в паху.
- Неспецифические симптомы патологии — одышка, бледность кожи, гипергидроз, «жар» в теле, «приливы», покраснение шеи, кардиалгия, аритмия, гипертензия, сменяющаяся гипотензией, нарушение сна и аппетита, депрессивные признаки, отсутствие интереса к жизни, гипотонус кожи.
- Возникают боли в костях в результате остеопороза.
Инволюция тестикул сопровождается потерей фертильности. У молодых мужчин 20-25 лет присутствуют редкие поллюции, эрекция, сохраняется половое влечение.
Ближе к 30 годам андрогенный дефицит становится максимально выраженным, что проявляется снижением либидо, уменьшением яркости оргазма и развитием импотенции. Взрослые пациенты часто становятся алкоголиками, наркоманами, гомосексуалами, особенно в условиях стресса.
Ослабление полового влечения, нарушение половой функции и бесплодие – наиболее частые признаки, с которыми больные люди приходят к врачу.
Лица с синдромом Клайнфельтера часто страдают коллагенозами, гипо- или гипертиреозом, заболеваниями глаз, имеют аномалии скелета и пороки сердца. Если лечение патологии не начать вовремя, могут развиться тяжелые осложнения и печальные последствия.
У больных нарушается психо-эмоциональное состояние, формируется умственная отсталость, появляются суицидальные наклонности, злоупотребление крепкими спиртными напитками, нарушается толерантность к глюкозе, кости становятся хрупкими, усугубляются врожденные пороки сердца, появляются новообразования молочных желез.
Многие случаи синдрома Клайнфельтера. остаются недиагностированными. Это приводит к отсутствию лечения, снижению качества жизни, инвалидизации больных, развитию остеопороза и сердечно-сосудистых заболеваний.
Пренатальная диагностика
Инвазивная пренатальная диагностика и последующее кариотипирование позволяют поставить правильный диагноз.
Существует две методики, с помощью которых можно получить материал для исследования:
- Биопсию хориона проводят с 9,5 по 12 неделю беременности.
- Амниоцентез – с 16 по 18 неделю.
Каждый из этих методов может выявить хромосомные аномалии с точностью до 99%.
Во время диагностического хирургического вмешательства получают ткани плода, из которых извлекают ДНК будущего ребенка. В лаборатории генетический материал исследуют на наличие хромосомных патологий.
- При биопсии специалисты прокалывают пункционной иглой переднюю брюшную стенку беременной женщины и извлекают ворсинки хориона с плаценты. Процедура контролируется аппаратом УЗИ.
- Амниоцентез — забор амниотической жидкости, содержащей генетическую информацию. Специальную иглу вводят в полость матки под контролем датчика аппарата УЗИ и собирают околоплодные воды.
Постнатальная диагностика
Диагностикой генетических заболеваний в постнатальном периоде занимаются эндокринологи, андрологи и генетики.
Специалисты начинают свою работу со сбора жалоб, анамнеза жизни и болезни. Они выясняют: время появления симптоматики, ее изменение, случаи генетических недугов в семье и в ближайшем поколении.
Затем переходят к визуальному осмотру, проведению при необходимости пальпации, перкуссии и аускультации. Скудная клиническая картина синдрома Клайнфельтера не всегда позволяет вовремя диагностировать патологию и начать заместительную гормонотерапию.
Диагностическим признаком заболевания являются тельца Барра, которые обнаруживают в клетках слизистой оболочки рта.Исследование кариотипа позволяет поставить окончательный диагноз. Кариотипирование проводится всем бесплодным мужчинам с гинекомастией и мальчикам с умственной отсталостью.
Дополнительные диагностические методы:
- Ультразвуковое исследование мошонки позволяет определить размеры и структуру яичек.
- Ультразвуковое исследование сердца проводится с целью обнаружения врожденных пороков.
- Денситометрия — метод выявления остеопороза.
- Определение в крови половых гормонов — тестостерона, ФСГ и ЛГ.
- Спермограмма — анализ эякулята, проводимый с целью определения фертильности мужчины, наличия половых заболеваний, количества и активности сперматозоидов. У лиц с синдромом Клайнфельтера отмечается снижение количества или полное отсутствие сперматозоидов в эякуляте.
- Биопсия яичек выявляет состояние сперматогенеза и имеет большую диагностическую ценность.
Гормонотерапия
Заместительная гормонотерапия проводится пожизненно и заключается в назначении препаратов – синтетических аналогов тестостерона, которые вводятся внутримышечно, перорально или сублингвально. «Тестостерона пропионат», «Тестостерона энантат», «Сустанона-250», «Метилтестостерон» являются препаратами выбора.
Наиболее популярны инъекционные формы тестостерона. Дозу лекарственного средства подбирает врач индивидуально каждому пациенту. При этом необходим постоянный контроль уровня тестостерона в крови.
Своевременно начатое и адекватное лечение придает фигуре мужеподобность, улучшает внешний вид и общее самочувствие больного, возвращает его к полноценной жизни.
- «Тестостерона пропионат» необходимо вводить каждые 2-3 дня. Такая монотерапия практически не проводится.
- «Тестостерона ципионат» – эфир пролонгированного действия, который необходимо использовать каждые 7-14 дней.
- Комбинированные препараты содержат смесь нескольких эфиров тестостерона. Они являются очень популярными в нашей стране. Вводят их также внутримышечно. «Тестостерона капронат» имеет продолжительное действие — до месяца.
- В настоящее время фармакологическая промышленность разработала микрокапсулированные лекарственные формы гормона, которые действуют три месяца. Такие препараты имеют существенный недостаток: они вызывают значительные колебания тестостерона в крови, что негативно сказывается на самочувствии больных.
Симптоматическое лечение
- При необходимости больным назначают препараты, укрепляющие костную ткань, снижающие кровяное давление, нормализующие уровень глюкозы в крови.
- При умственной отсталости используют нейрометаболики и психостимуляторы.
- В подростковом возрасте назначаются пластыри на основе тестостерона.
- Увеличенные молочные железы удаляют операционным путем. Больным проводят мастэктомию. Криптохизм лечат также с помощью хирургического вмешательства. Операцию проводят детям в возрасте 1-1,5 лет. Неопущение яичка приводит к патологическим процессам в тканях семенных желез и развитию необратимого бесплодия.
- Психотерапия показана всем больным с целью повышения трудоспособности и социальной адаптации.
- Детям назначают занятия ЛФК, коррекционные занятия с логопедом, закаливание.
Современные методы экстракорпорального оплодотворения позволяют больным с синдромом Клайнфельтера иметь детей. При этом забор генетического материала осуществляют непосредственно из яичка путем его биопсии. Сперматозоидами оплодотворяют яйцеклетки и получают здоровое потомство.
Донорская сперма — еще один шанс завести ребенка в семьях, где партнер болен.
Прогноз
Прогноз для жизни в целом благоприятный, а для восстановления способности к оплодотворению — крайне отрицательный. Больные способны вести нормальную жизнь, отдыхать и трудиться. Адаптироваться им в современном обществе поможет ранняя гормональная терапии и психотерапия.
Специфической профилактики заболевания не существует. Медико-генетическое консультирование проводится в обязательном порядке тем лицам, у которых в предыдущих поколениях регистрировались наследственные недуги. Правильное питание позволяет нормализовать массу тела и предупредить развитие ожирения и сахарного диабета.
: о синдроме Клайнфельтера
Источник: https://sindrom.info/klajnfeltera/
Синдром Клайнфельтера: мужчина с женской хромосомой

Елена Шведкина об одном из самых распространенных генетических заболеваний — больные жалуются на бесплодие, эректильную дисфункцию, гинекомастию и остеопороз
Синдром Клайнфельтера — генетическое заболевание, характеризующееся дополнительной женской половой хромосомой Х (одной или даже несколькими) в мужском кариотипе ХY. При этом в мужских половых железах — яичках — образуется недостаточно половых гормонов.
Как известно, генетический набор человека насчитывает 46 хромосом, из которых 22 пары называются соматическими, а 23‑я пара — половая.
Женщины имеют пару половых хромосом ХХ, а мужчины — ХY.
Для синдрома Клайнфельтера обязательно наличие мужской Y-хромосомы, поэтому, несмотря на дополнительные Х-хромосомы, пациенты всегда являются мужчинами.
Классификация: виды кариотипов при синдроме Клайнфельтера
По количеству дополнительных Х-хромосом различают следующие варианты синдрома Клайнфельтера:
- 47,ХХY — наиболее часто встречающийся
- 48,ХХХY
- 49,ХХХХY
Кроме того, к синдрому Клайнфельтера также относят мужские кариотипы, включающие, помимо дополнительных Х-хромосом, дополнительную Y-хромосому — 48,ХХYY. И, наконец, среди пациентов с этим синдромом встречаются лица с мозаичным кариотипом 46,ХY/47,ХХY (то есть часть клеток имеет нормальный хромосомный набор).
История открытия синдрома
Синдром получил свое название в честь Гарри Клайнфельтера — врача, в 1942 году впервые описавшего клиническую картину болезни. Клайнфельтер с коллегами опубликовали отчет об обследовании 9 мужчин, объединенных общими симптомами, такими как слабое оволосение тела, евнухоидный тип телосложения, высокий рост и уменьшенные в размерах яички.
Позднее, в 1956 г., генетики Планкетт и Барр (Е. R. Plankett, М. L. Barr) обнаружили у мужчин с синдромом Клайнфельтера тельца полового хроматина в ядрах клеток слизистой оболочки полости рта, а в 1959 году Полани и Форд (P. E. Polanyi, S. E.
Ford) с сотрудниками показали, что у больных в хромосомном наборе имеется лишняя Х-хромосома.
Активные исследования данной патологии велись в 70‑х годах в США. Тогда всех новорожденных мальчиков подвергали кариотипированию, в результате чего удалось достоверно выявить распространенность и генетические особенности синдрома Клайнфельтера.
Любопытно, что мыши также могут иметь синдром трисомии по половым хромосомам XXY, что позволяет эффективно использовать их в качестве моделей для исследования синдрома Клайнфельтера.
Этиология и причины нарушения
Синдром Клайнфельтера относится к генетическим заболеваниям, не передающимся по наследству, поскольку больные, за редким исключением, бесплодны.
Патология, как правило, возникает в результате нарушения расхождения хромосом на ранних стадиях формирования яйцеклеток и сперматозоидов. При этом синдром Клайнфельтера, возникающий за счет нарушения в женских половых клетках, встречается в три раза чаще.
Мозаичные формы обусловлены патологией деления клеток на ранних стадиях эмбриогенеза, поэтому часть клеток у таких пациентов имеет нормальный кариотип.Причины нерасхождения половых хромосом и нарушения деления клеток на самых ранних стадиях эмбриогенеза до сих пор малоизучены. В отличие от других хромосомных заболеваний, влияние возраста родителей отсутствует или выражено незначительно.
Ранние признаки
В отличие от большинства заболеваний, связанных с нарушением количества хромосом, внутриутробное развитие детей с синдромом Клайнфельтера проходит нормально, склонности к преждевременному прерыванию беременности не наблюдается.
Так что в младенческом и раннем детском возрасте заподозрить патологию практически невозможно. Более того, клинические признаки классического синдрома Клайнфельтера проявляются, как правило, только в подростковом периоде.
Однако есть симптомы, которые позволяют заподозрить наличие синдрома Клайнфельтера в препубертатном периоде:
- высокий рост (пик прибавки роста приходится на период между 5–8 годами);
- длинные ноги (непропорциональное телосложение);
- высокая талия.
У части пациентов наблюдается некоторая задержка в развитии речи.
В подростковом возрасте синдром часто проявляется гинекомастией, которая при данной патологии имеет вид двустороннего симметричного безболезненного увеличения грудных желез. Так как такого рода гинекомастия часто наблюдается у совершенно здоровых подростков, этот симптом часто остается без внимания.
В норме подростковая гинекомастия бесследно исчезает в течение нескольких лет, у пациентов же с синдромом Клайнфельтера обратной инволюции грудных желез не происходит.
В некоторых случаях гинекомастия может не развиваться вовсе, и тогда патология проявляется признаками андрогенной недостаточности уже в постпубертатный период.
Симптомы андрогенной недостаточности при синдроме Клайнфельтера
Андрогенная недостаточность при синдроме Клайнфельтера связана с постепенной атрофией яичек, что приводит к снижению синтеза тестостерона. Степень недостаточности андрогенов резко варьирует.
В первую очередь обращают на себя внимание внешние признаки гипогонадизма:
- скудная растительность на лице или же полное ее отсутствие;
- рост волос на лобке по женскому типу;
- волосы на груди и других частях тела отсутствуют;
- маленький объем яичек (2–4 мл) и их плотная консистенция (патогномоничный признак).
Поскольку дегенерация половых желез, как правило, развивается в постпубертатный период, у большинства пациентов размеры мужских половых органов, за исключением яичек, соответствуют возрастным нормам.
Пациенты могут жаловаться на ослабление либидо и снижение потенции.
У многих мужчин с синдромом Клайнфельтера половое влечение вовсе не возникает, а некоторые — напротив, заводят семью и живут нормальной половой жизнью.
Наиболее постоянный признак патологии — бесплодие, именно оно чаще всего становится причиной обращения таких пациентов к врачу. У 10 % мужчин с азооспемией обнаруживают синдром Клайнфельтера.
Всем пациентам с нарушениями сперматогенеза необходимо определять кариотип для исключения или подтверждения диагноза синдрома Клайнфельтера.
Недостаток андрогенов приводит к развитию остеопороза, анемии и слабости скелетной мускулатуры. У трети больных можно наблюдать варикозное расширение вен голеней.
Андрогены влияют на обмен веществ, поэтому больные с синдромом Клайнфельтера склонны к ожирению, нарушению толерантности к глюкозе и сахарному диабету второго типа.
Доказана предрасположенность таких пациентов к аутоиммунным заболеваниям (ревматоидный артрит, системная красная волчанка, аутоиммунные заболевания щитовидной железы и другие).
Психологические особенности
Коэффициент интеллекта у больных с классическим синдромом Клайнфельтера варьирует от значений ниже среднего до показателей, значительно превышающих средний уровень.
Однако во всех случаях отмечается диспропорция между общим уровнем интеллекта и вербальными способностями, так что нередко пациенты с достаточно высоким IQ испытывают трудности при восприятии больших объемов материала на слух, а также при построении фраз, содержащих сложные грамматические конструкции.
Такие особенности причиняют пациентам много неприятностей в период обучения и нередко продолжают сказываться на профессиональной деятельности.
Данные о психологических особенностях больных с синдромом Клайнфельтера достаточно противоречивы, однако большинство специалистов оценивают пациентов как скромных, робких людей с несколько заниженной самооценкой и повышенной чувствительностью.Есть данные, свидетельствующие о склонности пациентов с синдромом Клайнфельтера к гомосексуализму, алкоголизму и наркомании.
Сложно сказать, вызваны ли особенности психики у таких больных непосредственным влиянием хромосомной аномалии, или же это реакция на проблемы в сексуальной сфере.
В отношении разных цитогенетических вариантов синдрома Клайнфельтера справедливо правило, что с увеличением количества дополнительных Х-хромосом увеличивается количество и выраженность патологических симптомов.
Диагностика синдрома Клайнфельтера
Во многих странах синдром Клайнфельтера часто диагностируется ещё до рождения ребёнка, так как многие женщины позднего детородного возраста, в связи с высоким риском генетических дефектов у будущего потомства, используют пренатальную генетическую диагностику плода. Нередко пренатальное выявление синдрома Клайнфельтера является поводом для прерывания беременности, в том числе и по рекомендации врачей. В России анализ кариотипа будущего ребёнка проводится крайне редко.
При подозрении на синдром Клайнфельтера проводят лабораторный анализ крови для определения уровня мужских половых гормонов. Необходима дифференциальная диагностика с другими заболеваниями, протекающими с проявлениями андрогенной недостаточности. Точный диагноз синдрома Клайнфельтера ставят на основании изучения кариотипа (набора хромосом) больного.
Исследования, необходимые для подтверждения диагноза
|
Анализы |
Результаты |
|
Кариотип |
47,ХХY (80 % случаев) 48,ХХYY 48,ХХХY 49,ХХХХY 46,ХY/47,ХХY |
|
Концентрация ЛГ, ФСГ |
Повышена, особенно ФСГ |
|
Концентрация общего тестостерона |
Чаще снижена (в некоторых случаях нормальная за счет повышения секс-стероид-связывающего глобулина СССГ или на начальной стадии развития заболевания) |
У всех мужчин с резко повышенными концентрациями гонадотропинов необходимо исключить синдром Клайнфельтера, так как нередко первый лабораторный признак этой генетической патологии — повышение в крови концентрации гонадотропинов при нормальном содержании общего тестостерона.
Синдром Клайнфельтера необходимо дифференцировать от других форм первичного гипогонадизма. В любом случае при повышении уровня ФСГ в крови необходимо определение кариотипа для исключения в первую очередь синдрома Клайнфельтера.
Цели лечения синдрома Клайнфельтера:
- Восстановление нормального содержания тестостерона
- Восстановление сексуальной функции
- Ликвидация метаболических нарушений
При клинически выраженной патологии необходима пожизненная заместительная терапия препаратами тестостерона.
Адекватная терапия позволяет не только улучшить внешний вид и общее самочувствие больного, но и вернуть способность к нормальной половой жизни. Кроме того, заместительная терапия предупреждает развитие остеопороза, купирует мышечную слабость. В юном возрасте лечение необходимо начинать сразу же после постановки диагноза.
При синдроме Клайнфельтера лучше использовать препараты тестостерона длительного действия:
- смесь эфиров тестостерона в виде масляного раствора, инъекции которого необходимо делать 2–3 раза в месяц;
- тестостерона ундеканоат в виде масляного раствора — препарат-депо с замедленным высвобождением действующего вещества — инъекции 1 раз в 3 месяца.
Гормонолечение при наличии Х хромосомы у мужчин должно носить постоянный характер. Дозу препарата подбирают индивидуально под контролем уровня тестостерона и ЛГ в сыворотке крови.
Уже развившаяся гинекомастия при синдроме Клайнфельтера не подвергается инволюции даже в случае адекватного лечения, поэтому часто приходится прибегать к хирургической коррекции (мастэктомии).
Для профилактики таких сопутствующих заболеваний, как ожирение и сахарный диабет второго типа, больным рекомендуют придерживаться диеты и следить за собственным весом.
Мониторинг пациентов с синдромом Клайнфельтера следует осуществлять не реже 1 раза в 6–12 месяцев. Он должен включать следующие исследования:
- общий анализ крови для оценки уровня гемоглобина и гематокрита;
- гормональный анализ крови, включающий определение тестостерона и ЛГ (проводится на фоне лекарственной терапии за 1–2 дня до очередной инъекции тестостерона);
- денситометрию (всем пациентам, у которых на момент постановки диагноза были обнаружены остеопения или остеопороз).
Внедрение интрацитоплазматической инъекции сперматозоида в яйцеклетку (ИКСИ) и данные о возможности присутствия зародышевых клеток в яичках у пациентов с синдромом Клайнфельтера предопределили применение метода искусственного оплодотворения для данной категории пациентов, некоторые попытки были удачными.