
Синдром Элерса-данлоса
Содержание
Синдром Элерса-Данлоса: что это такое, опасные симптомы и современное лечение
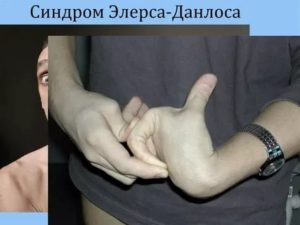
Синдром Элерса — Данлоса — это группа наследственных системных заболеваний соединительной ткани, вызванных дефектом в синтезе коллагена. В зависимости от отдельной мутации, серьёзность синдрома может измениться от умеренного до опасного для жизни. Лечения нет, но существует терапия (уход), смягчающая последствия.
Синдром Элерса-Данлоса был назван в честь своих первооткрывателей — дерматологов из Дании и Франции Э. Элерса и А. Данлоса. Они первые описали причины и симптомы данной патологии.
В настоящее время существует 6 основных форм синдрома Элерса-Данлоса, которые отличаются симптоматикой, способом наследования, биологическими и химическими изменениями в тканях.
Среди всех существующих типов дисплазий этот синдром считается самым распространенным.
Причины синдрома
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
- Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена.
- Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа;
- При VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена;
- VII тип обусловлен нарушением превращения проколлагена I типа в коллаген;
- X тип — патологией плазменного фибронектина, участвующего в организации межклеточного матрикса и т. п.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Причины
В разное время изучения наследственной патологии синдрома Элерса-Данлоса существовала классификация, по которой выделяли от трёх до одиннадцати типов заболевания. По мере того как накапливались молекулярно-генетические данные, появилась необходимость в пересмотре прежних вариантов подразделений синдрома Элерса-Данлоса.
Поэтому ещё в 1997 году учёные-медики классифицировали это заболевание на десять типов и на данный момент она абсолютно приближена к причинам возникновения аномалии. Синдром Элерса-Данлоса имеет разные типы наследования для каждого типа.
Аутосомно-доминантное характерно для первого, второго, третьего, четвёртого, седьмого и восьмого, а аутосомно-рецессивное наследование – для шестого, Х-сцепленное – для пятого и девятого, а десятый не имеет установленного типа наследования, так как встречается довольно редко. Это заболевание является примером разной локусной гетерогенности.
Именно эти локусы, в которых происходят аномальные изменения вызывающие синдром Элерса-Данлоса, имеют непосредственное отношение к образованию белков соединительной ткани (коллагена). Эти волокна из коллагена, в результате мутаций, становятся неправильной формы и располагаются неупорядоченно.Кроме мутированных генов, причиной в возникновении синдрома Элерса-Данлоса являются мутации в тех генах, в которых локализуются белки с экстраклеточным матриксом, и среди них выделяют – тенасцин Х, люмикан и декорин.
Формы
Первая классификация синдрома Элерса- Данлоса выделяла более 10 типов заболевания, но в 1997 году была предложена более простая классификация, сокращающая число основных типов до 6. Не вошедшие в классификацию типы либо наблюдаются исключительно в отдельных семьях (типы V, VIII и X), либо недостаточно характеризованы (типы IX и XI).
Для каждого из выделенных типов идентифицированы вызывающие заболевание мутации (исключение — тип гиперподвижности), которые благодаря генетическому анализу позволяют точно определить тип синдрома, отличающегося значительной вариативностью признаков.
В современной классификации для каждого типа заболевания используются описательные имена. Выделяют:
- Классический тип, для которого характерна повышенная подвижность и деформация суставов, вялость кожных покровов. Синдром Элерса – Данлоса у детей при данном типе заболевания часто сопровождается сколиозом. Предыдущая классификация относила данный тип к 1 и 2 типу заболевания.
- Гиперподвижный тип, который отличается гиперподвижностью суставов, хроническими мышечно-скелетными болями и незначительными поражениями кожи. В предыдущей классификации относился к 3 типу.
- Сосудистый тип, который проявляется в повышенной хрупкости сосудов и спонтанных разрывах артерий. Сопровождается частыми внутренними кровотечениями, поэтому представляет угрозу для жизни больного. Первая классификация относила сосудистый тип к 4 типу заболевания.
- Кифосколиоз – редко встречающийся тип, для которого характерно наличие деформации позвоночника (кифоз и сколиоз). Часто наблюдаются миопия, разрывы роговицы и другие изменения в глазном яблоке, мышечная гипотония. Согласно первой классификации, относится к 6 типу.
- Артрохалазия, проявляющаяся повышенной мобильностью суставов, гиперрастяжимой кожей и маленьким ростом больного. По первой классификации относилась к типам 7A и B.
- Дерматоспараксис – крайне редкий тип, который отличается мягкостью, вялостью и обвисанием кожи. Согласно первой классификации относится к типу 7C.
- Также отдельные исследователи выделяют тип недостатка Тинасцина-Х, который отличается хрупкостью тканей и признаками, свойственными классическому типу заболевания.
Симптомы синдрома
В связи с тем, что соединительнотканные волокна имеются практически в каждом органе, клинические проявления синдрома Элерса-Данлоса полиморфны и носят генерализованный характер.
Кожа при данном заболевании нежная и бархатистая на ощупь, морщинистая (особенно на ладонях и стопах), слабо связанная с подлежащими тканями и гиперэластичная, гиперрастяжимая, то есть легко оттягивается при формировании складки.
Однако с увеличением возраста больного выраженность данных характеристик кожи снижается.
Минимальная травматизация столь ранимой и хрупкой кожи приводит к возникновению длительно не заживающих ран, после заживления которых могут оставаться атрофичные или келоидные рубцы, а также псевдоопухоли.
Костные проявления данного заболевания представлены деформацией грудной клетки (килевидной или воронкообразной), позвоночного столба (кифозы, сколиозы), частичным отсутствием зубов, неправильным прикусом и косолапостью.
Поражение суставов характеризуется их гипермобильностью, что нередко приводит к вывихам и подвывихам.
Нередко у больных синдромом Элерса-Данлоса обнаруживается патология сердечно-сосудистой системы (врождённые пороки сердца, варикозное расширение сосудов, аневризмы сосудов головного мозга, склонность к кровотечениям).
Со стороны внутренних органов нередкой патологией являются рецидивирующий спонтанный пневмоторакс, диафрагмальные грыжи, дивертикулы кишечника, паховые и пупочные грыжи, птозы внутренних органов (их опущение).Офтальмологическая патология при синдроме гиперэластичности кожи представлена, как правило, миопией, косоглазием, птозом век, спонтанной отслойкой сетчатки и разрывами глазного яблока и роговицы при малейших повреждениях.
Диагностика
Диагностика синдрома Элерса-Данлоса основана на данных:
- Анамнеза заболевания и генеалогических данных.
- Обследования, во время которого изучают состояние опорно-двигательного аппарата пациента, оценивают состояние кожи (основной признак синдрома Элерса — Данлоса — повышенная растяжимость кожи, которую можно оттянуть на 1,5—2 см в тех местах, где у здорового человека это сделать невозможно) и подвижность суставов.
- Биопсии кожи, которая позволяет провести гистологическое, гистохимическое и электронно-микроскопическое исследования, необходимые для уточнения диагноза при подозрении на дерматоспараксис или артрохалазию.
- Молекулярно-генетических исследований, которые проводятся при подозрении на синдром Элерса – Данлоса. Основание – наличие основных (гипермобильность суставов, повышенная эластичности кожи, склонность к кровотечениям) и дополнительных (патологии сосудов, сердца, глаз и т. д.) диагностических критериев.
Также проводятся:
- УЗИ внутренних органов, позволяющие выявить патологии, которые может вызвать недостаточность связочного аппарата;
- офтальмологические обследования, включающие биомикроскопию, фундус-графию (позволяет изучить состояние дна глазного яблока) и компьютерную томографию глазного яблока;
- эхокардиограмму, позволяющую выявить пролапсы клапанов или расширение аорты;
- тилт-тест (пассивную ортостатическую пробу) при наличии постуральной ортостатической тахикардии.
Лечение
Вылечить этот синдром, к сожалению, невозможно, терапию проводят для ликвидации клинической картины, а, кроме того, в целях поддержки деятельности внутренних органов. Чтобы стабилизировать общее состояние сердца, нервной и опорно-двигательной системы, а также суставов и кожи, используются следующие лекарственные препараты, составляющие основу лечения синдрома Элерса-Данлоса:
- Лечение витаминами группы В в сочетании с назначением пациентам аскорбиновой кислоты и токоферола.
- Медицинские препараты, которые активируют обменные процессы, к примеру, «Карнитин-хлорид».
- Назначение комплексов с минеральными веществами.
- Инъекционный ввод соматотропных гормонов.
- Применение глюкозамина – вещества, участвующего в продуцировании соединительнотканных элементов.
- Назначение лекарственного препарата «Остеокеа».
В результате приема данного средства возможно подержание состояния костных и нервных тканей, и мышечного аппарата в том числе.
Прогноз. Профилактика
Прогноз в большинстве случаев благоприятный, серьезнее он при первом (вследствие артропатий) и шестом (вследствие кровотечений и разрывов сосудов) типах. Детей следует ориентировать на выбор профессии, не связанной с физическими нагрузками, работой стоя.
Профилактика заключается в пренатальном выявлении заболевания у плода методом молекулярно-генетического анализа, прерывании беременности по медицинским показаниям. Постнатальная профилактика заболевания не разработана.
Профилактика осложнений включает в себя правильную и своевременную диагностику болезни и её лечение.
(1 5,00 из 5)
Загрузка…
Источник: https://osindromah.ru/geneticheskie/sindrom-elersa-danlosa.html
Синдрома Элерса-Данлоса
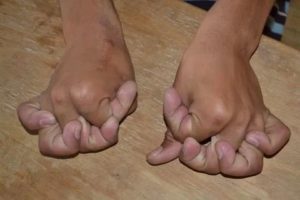
В этой статье мы расскажем о такой патологии, как синдром Элерса-Данлоса, что это такое, рассмотрим симптомы (фото больных), причины возникновения и метод терапии.
Что такое синдром Элерса-Данлоса
Синдром Элерса-Данлоса – это наследственное заболевание, спровоцированное дефектом синтеза коллагена. При такой заболевании наблюдается неправильное развитие соединительных тканей и гиперэластичность кожи.
Синдром имеет несколько типов, которые могут характеризоваться неестественной растяжимостью кожи, чрезмерной способностью суставов разгибаться в разные стороны, косоглазием, деформацией скелета.
Болезнь затрагивает большое количество систем организма и представляет интерес не только с точки зрения генетики, но и кардиологии, ортопедии, стоматологии, офтальмологии и других медицинских дисциплин.
Так же синдром Элерса-Данлоса отличается сложностью в диагностике из-за наличия легких форм недуга, поэтому нельзя с точностью оценить его распространенность в мире.
Эпидемиология
Точная распространенность и годовая заболеваемость неизвестны, оценки распространенности варьируются от 1/5 до 000-1/20000. Из-за клинической изменчивости эти оценки могут быть слишком низкими. Больше всего болезнь затрагивает – женский пол.
Причины синдрома Элерса-Данлоса
Синдром Элерса-Данлоса имеет несколько вариантов патологии, отличающихся друг от друга по типу наследования. Но их объединяет то , что в их основе лежит структурное либо количественное нарушение белка коллагена. В нынешнее время не для каждого типа заболевания найдены молекулярные механизмы синдрома.
Для первого типа синдрома характерна пониженная активность тех клеток соединительной ткани, отвечающие за синтез межклеточного матрикса. Так же происходит увеличение синтеза протеогликанов, а еще отсутствие ферментов, отвечающих за стабильное воссоздание белков коллагена.
При четвертом типе синдром Элерса-Данлоса наблюдается дефицит коллагена 3 типа. Шестой тип характеризуется недостатком биологического катализатора лизилгидроксилазы, который принимает участие в гидроксилировании лизина в молекулах проколлагена. Седьмой тип заболевания возникает вследствие изменения проколлагена первого типа, который становиться коллагеном.Если охарактеризовать заболевание в целом, в различных формах, то заболевание возникает вследствие снижения плотности и расстройство ориентации коллагеновых волокон, утончение соединительного отдела кожи, расширение кровеносных сосудов и большую видимость их на поверхности тела.
Симптомы синдрома Элерса-Данлоса
Симптомы могут возникать в любом возрасте, но их трудно диагностировать у маленьких детей из-за повышенной гипервозбудимости суставов. Клинические проявления очень разные. Первичное проявление – это это гиперэластичность различных суставов и аномальная эластичность кожного покрова (см. фото выше).
На ощупь кожа больного нежная, но на ступнях и ладонях присутствуют многочисленные морщины. При синдроме Элерса-Данлоса неестественная эластичность кожи наблюдается с ранних лет жизни человека, так же с возрастом характерно снижение такого отклонения.
Повреждения на коже людей, подверженных синдрому, ощущаются сильней и заживают медленно, а на их месте образуются следы в виде шрамов и опухолей.
Кроме гиперэластичности кожи у подверженных синдрому Элерса-Данлоса наблюдается сильная гиперэластичность суставов, как отдельной части тела, так и всего организма.
Суставы становятся невероятно гибкими и гнуться под неестественными им углами с того момента, как человек начинает учиться ходить, что нередко приводит к травмам. С возрастом ненормальная способность суставов к сгибанию ослабевает.
Повреждение скелета при синдроме представляет собой деформацию грудной клетки в виде дуги или полукруга.
В результате заболевания возникает сколиоз, косолапость. Со скелетом нарушается и расположение внутренних органов, опущение, наличие различных видов грыж, дивертикулез кишечника, пневмоторакс, ввиду нарушения целостности плевры.
Наличие синдрома ведет к патологиям зрения, среди которых встречаются:
- миопия;
- спонтанная отслойка сетчатки;
- косоглазие;
- разрыв глазного яблока и роговицы.
Что касается кардиологии, то у детей с синдромом Элерса-Данлоса возникают частые кровотечения и гематомы.
Не исключен порок сердца, варикоз, пролапс митрального клапана и аневризмы сосудов головного мозга. Нарушений умственного развития детей с данным заболеванием обычно не наблюдается.
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.
5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Диагностика синдрома Элерса-Данлоса
Диагностику синдрома Элерса-Данлоса лучше доверять опытному человеку, специализирующемуся именно в области этого заболевания.
Малознакомому с Элерсом-Данлосом врачу будет сложно выявить недуг и тем более его особенности. Это требование связанно с разнообразием индивидуальных проявлений такого заболевания у каждого пациента.
Существует множество разновидностей болезни, но есть характерные черты ее проявления.
Неестественная эластичность кожного покрова выявляется путем оттягивания кожи до того момента, пока не почувствуется сопротивление. Участки для осуществления проверки следует выбирать максимально нейтральные, где нормальная оттягиваемость кожи превышает полтора сантиметра.
Среди характерных для болезни синдромов присутствует гипермобильность суставов. Такой симптом определяется по шкале Бейтона, где 5 баллов означает отклонение от нормы. Этот симптом, с возрастом, наблюдается в меньшей степени.
О хрупкости суставных соединений свидетельствуют спонтанно возникающие синяки. Вместе с этим возможны долговременные кровотечения.
Даже при совсем слабых внешних воздействиях появляются различные травмы , которые оставляют атрофические следы ранений в виде шрамов и расширяются с течением времени. С ами ранения заживают дольше, чем у здоровых людей.
Характерной патологией синдрома Элерса-Данлоса является пролапс митрального клапана. Этот недуг выявляется проведением УЗИ сердца.Постоянная боль в области суставов и конечностей тела – еще одно проявление синдрома Элерса-Данлоса. При таком признаке рентген может не выявить отклонений, а пациент не всегда понимает, из какой части тела исходят болевые ощущения.
Из шкалы Брайтона берутся формальные критерии для того, чтобы диагностировать синдром Элерса-Данлоса. Первоначально функцией шкалы Бейтона была диагностика гипермобильности в суставах, но современные опыты показывают, что синдром гипермобильности суставов и повышенная сгибаемость суставов при Элерсе-Данлосе, – это аналогичные заболевания.
Шкала Бейтона
Основные критерии:
- 4+ баллов по шкале Бейтона выявленных в настоящее время, либо раньше;
- артралгия, длящаяся на протяжении 3 месяцев и более в 4 или более суставах.
Второстепенные критерии:
- 1-3 Балла по шкале Бейтона ( и 0 если вы старше 50-ти);
- артралгия в 1, 2 или 3 суставах с присутствием болезненных ощущений в спине;
- вывихи в одном и более суставах, либо в одном суставе несколько раз;
- более 3 воспалений мягких тканей, ревматизм;
- марфаноидный тип внешности (чрезмерная худощавость, высокий рост, арахнодактилия);
- необычные шрамы и ненормальная растяжимость и тонкость кожи;
- растянутая кожа век или миопия;
- ректальный пролапс, варикозное расширение вен, пролапс матки, грыжа.
Для того чтобы поставить диагноз “синдром Элерса-Данлоса” нужно выявить оба основных критерия, либо 1 из основных и 2 любых второстепенных, для постановки диагноза подойдет и наличие 4 второстепенных критериев.
Когда у вашего первостепенного родственника выявлен данный синдром, вам достаточно будет определить наличие 2 второстепенных критериев.
Лечение синдрома Элерса-Данлоса
Для эффективного устранения синдрома Элерса-Данлоса специфическая терапия до сих пор не разработана, но существуют методы лечения симптомов данной болезни.
Медикаментозная терапия
Для того, чтобы стабилизировать работу сердечно-сосудистой и нервной системы, а так же привести в норму работу суставов, опорно-двигательный аппарат, целостность и эластичность кожного покрова используют несколько вариантов лечения:
- лекарственные препараты в виде аскорбиновой кислоты, А, Е, В витаминов;
- комплексы минералов;
- инъекции соматотропного гормона для стимуляции роста;
- стимулирующие обмен веществ и регенерацию кожного покрова метаболические средства, такие как карнитин-хлорид;
- для стимуляции мозговой деятельности применяются нейрометаболические стимуляторы;
- для поддержания целостности скелета и соединительных тканей используются остеокеа или остеогенон;
- заживлению кожи способствуют такие препараты, как инозин, АТФ, коэнзим Q10.
- используется принимающий участие в синтезе хрящевых и в соединительных тканей глюкозамин.
Хирургическое вмешательство
К хирургическому вмешательству при синдроме Элерса-Данлоса стоит прибегать, только если возникают осложнения, угрожающие жизни. Перед хирургическими операциями должны быть проведены тщательные инвазивные диагностические процедуры для того чтобы оценить степень угрозы и необходимость хирургического вмешательства.
Среди хирургического лечения данного синдрома может быть необходимым: удаление псевдоопухолей, коррекция ВПС, реконструкция грудной стенки и т.д.
Дополнительные и альтернативные методы лечения, в том числе и в домашних условия
Среди процедур назначаются:
- лечебная физкультура и массаж
- рефлексотерапия (воздействие на биологически активные точки организма, отвечающие за работу различных его систем)
- различные физиотерапевтические процедуры (к примеру, лазерная акупунктура)
Также назначается диета с повышенным потреблением белка. Такая диета содержит потребление костных бульонов и заливных блюд.
Прогноз для больных
Повышенного риска ранней смертности нет, однако, в связи с нестабильностью суставов, хроническим и острым болевым синдромом, и симптомами проявляющееся вне костно-мышечной системы, качество жизни больного серьезно ухудшается.
Как уже говорилось выше, специального лечения нет. Необходимо проходить индивидуализированные вспомогательные и симптоматические методы лечения включающие физиотерапию, реабилитацию, прием анальгетиков и соответствующую терапию внесуставных симптомов. Хирургические меры следует рассматривать с умом.
Источник: https://tvojajbolit.ru/pediatriya/sindroma-elersa-danlosa/
Синдром Элерса-Данло у детей. Клинические рекомендации

- Коллаген
- Гипермобильность
- Гиперрастяжимость
- Геморрагический синдром
- Пролапс митрального клапана
СЭД-синдром Элерса-Данло/Данлоса
УЗИ-ультразвуковое исследование
МРТ-магнитно-ядерная томография
КТ— компьютерная томографию
КФК-креатинин-фосфокиназа.
Термины и определения
Коллаген — фибриллярный белок, составляющий основу соединительной ткани организма (сухожилие, кость, хрящ, дерма и т. п.) и обеспечивающий её прочность и эластичность.
Тилт-тест -пассивная ортостатическая проба.
Адентия – полное или частичное отсутствие зубов, возникающее вследствие их потери или аномалии развития зубочелюстной системы.
1.1 Определение
Синдром Элерса—Данло/Данлоса, или СЭД, — это наследственная мезенхимальная дисплазия, гетерогенная группа наследственных заболеваний с проявлениями со стороны кожи, опорно-двигательного аппарата и других органов.
Болезнь развивается в связи с дефектами молекулярной структуры коллагена, поражая соединительную ткань организма и формируя симптомокомплекс, также известный как «гиперэластичность кожи», «Cutis hyperelastica», несовершенный десмогенез Русакова, синдром Черногубова—Элерса-Данлоса.
Заболевание впервые упоминается в 1682 г. J. van Meekeren; приоритет первого детального описания принадлежит А.Н. Черногубову (1891). Синдром же назван в честь двух дерматологов, идентифицировавших его в начале XX в.
: Эдварда Элерса (1863-1937) издании и Генри Александра Данло (1844-1912) из Франции, хотя эти авторы описали это заболевание намного позднее в 1901 и 1908 гг [1].
1.2 Этиология и патогенез
Основой патогенетических процессов являются дегенеративные изменения соединительной ткани, которые вызываются нарушением биосинтеза коллагена.
Коллаген является группой близкородственных фибриллярных белков (известно 19 типов), в состав которых входят остатки нестандартных аминокислот (на 3-гидроксипролин, 4-гидроксипролин и 5-гидроксилизин приходится около 21 % от общего числа остатков).
Синтез и созревание фибриллярных белков — сложный многоступенчатый процесс, включающий следующие пост трансляционные изменения:
- гидроксилирование при помощи фермента лизилгидроксилазы аминокислот пролина и лизина (необходимо для стабилизации тройной спирали коллагена) с последующим образованием нестандартных аминокислот гидроксипролина (Hyp) и гидроксилизина (Hyl);
- присоединение остатков сахаров к молекуле гидроксилизина (гликозилирование);
- частичное разложение белков коллагена (протеолиз), при котором под воздействием ферментов отщепляется «сигнальный» пептид, а также N- и С-концевые пропептиды; образование тройной спирали коллагена[2,3].
1.3 Эпидемиология
Полиморфизм клинических симптомов, множество мутаций, которые иногда регистрируются только в единичных семьях, отсутствие популяционных рандомизированных исследований не позволяет определять эпидемиологические частотные характеристики СЭД,
Наряду с возникновением спонтанных мутаций описаны случаи, для которых характерен аутосомно-доминантный тип наследования, иногда с передачей через X-хромосому.
Имеются изоляты с выраженным эффектом родоначальника на протяжении нескольких поколений, в которых больные с синдромом 1-го типа составляют 10% всего населения. Примером такого изолята является с.
Гобу Абшеронского района Азербайджана, где часто встречается синдром Элерса—Данло [4].
1.4 Кодирование по МКБ-10
Код по МКБ-10: Q79.6. Синдром Элерса-Данло.
1.5 Классификация
Наиболее распространенные формы, для которых расшифрован генетический дефект, следующие:
? (Hypermobility) СЭД, связан с геном COL3A1, collagen, type III, alpha 1; TNXB,tenascin XB, Fibronectin type III domain containing, обусловлен аутосомно-доминантным механизмом наследования.
Возникает в результате мутации любого из двух генов, которые вызывают Сосудистый тип и СЭД с дефицитом тенасцина-X, представлены гипермобильностью суставов, которая поражает 1 из 10000 до 15000 человек, вызывает нестабильность крупных и мелких суставов.
Признаки и симптомы могут быть не диагностированы (не признаны) врачами или, как правило, ошибочно диагностированы как фибромиалгия и обычно больным не ставят диагноз, пока не проявятся серьёзные осложнения. Пациенты часто испытывают боли в суставах и конечностях, им свойственно раннее начало остеопороза после 30 лет
? Классический (Classical) СЭД вызван геном COL5A1, collagen, type V, alpha 1 и другими генами семейства Collagens, затрагивает коллаген типа V, также коллаген типа I; поражает 1 из 20000 до 40000 человек, и характеризуется повышенной растяжимостью кожи, шрамы и раны, не заживают должным образом, геморрагический синдром имеет проявления сосудисто-тромбоцитарного типа кровоточивости, могут формироваться кисты под кожей, явления преждевременного старения, часто возникают доброкачественные новообразования кожи и подкожной клетчатки.
? Сосудистые формы (Vascular) СЭД вызваны аутосомно-доминантным дефектом гена COL3A1 в синтезе коллагена типа III, встречается у 1 из 250 000 человек. Этот тип считается весьма серьезным из-за риска профузных кровотечений из внутренних органов или разрывов кровеносных сосудов.
Люди с сосудистыми формами СЭД имеют очень тонкую кожу с просвечивающей кровеносной сетью. Высокие факторы риска, связанные с сосудистыми проявлениями СЭД часто приводит к снижению продолжительности жизни до 50 лет.Остальные три формы СЭД редки, будучи представлены всего приблизительно 100 случаями по всему миру.
? Ахондроплазиспластический тип (Arthrochalasis) СЭД, который характеризуется дефектом коллагена 1 типа за счет генов COL1A2 collagen, type I, alpha 2 и COL1A collagen, type I, alpha 1, при этом варианте ребенок может рождаться с врожденным вывихом бедра; было описано только около 30 случаев. Пациенты с этим типом СЭД имеют раннее начало артрита, частое появление на коже кровоподтеков, эластичную кожу и атрофические рубцы.
? Люди с дерматоспараксис (Dermatosparaxis) СЭД имеют дефектный ген ADAMTS2, ADAM metallopeptidase with thrombospondin type 1 motif, 2, о котором было сообщено в 10 случаях по всему миру, имеют чрезвычайно хрупкую кожу с мягкой, рыхлой текстурой.
Они тоже очень восприимчивы к кровоподтекам, но расстройство не препятствует заживлению ран, как это происходит при других формах заболевания. Растяжимость кожи вариабельна, и не характерна атрофичность рубцов.
Однако мышечные и суставные боли начинаются очень рано с изнуряющим и хроническим характером.
Наименее распространенной формой (Kyphoscoliosis) СЭД является кифосколиоз, о котором было сообщено лишь в нескольких случаях во всем мире.
Представляет собой аутосомно-доминантный дефект, вызывающий недостаток фермента, называемого лизин гидролазой за счет дефектного гена PLOD1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase Слабый мышечный тонус и задержка моторного развития, часто приводит к потере мобильности к 20-30-х годам, являются общими для этого типа СЭД. По мере того как болезнь прогрессирует, позвоночник становится все более изогнутым. Глаза, имеют небольшие роговицы, которые легко повреждаются и разрываются[5,6].
В связи с тем, что классификационные критерии находятся в стадии детализации можно оценить ее уровень достоверности 2, уровень доказательности В.
2.1 Жалобы и анамнез
Вариабельность клинических проявлений определяет характер жалоб пациентов. Часто, если у пациента нет грубых функциональных нарушений, то жалобы вовсе пациент/его родители не предъявляют.
Вместе с тем учитывая семейный характер заболевания диагностика, начинается практически с момента рождения: сниженный мышечный тонус, задержка формирования моторных навыков, выявление кардиологических симптомов, склонность к образованию грыж[7].
Изучение анамнеза является важным диагностическим приемом. Анализ отдельных симптомов в аспекте возможного наличия у пациента СЭД позволяет обеспечить комплексную диагностику и назначить более адекватный режим коррекции выявленных нарушений.
2.2 Физикальное обследование
Исследователи выделяют 10 вариантов СЭД. Шесть основных типов СЭД имеют свои специфические симптомы. Тяжесть этих симптомов вариабельна, иногда незначительный СЭД не диагностируется, а иногда приводит к серьезным проблемам мобильности. Большинство симптомов могут быть объединены в 2 группы: кожные и суставные.
Симптомы СЭД включают:
? гипермобильность суставов, что может привести к дислокации костей и хронической боли;
? нежную кожу, которая подвержена травмам, формированию аномальной рубцовой ткани;
? чрезмерную эластичность кожи, что делает кожу склонной к перерастяжению, уязвимой для повреждений и увеличивает риск повреждения внутренних органов при травмах;
? снижение сосудистого тонуса, приводящее к различным вариантам сосудистой недостаточности;
? глаза: птоз, отслойка сетчатки, остатки эпиканта, разрыв глазного яблока, периорбитальная полнота тканей за счет гиперэластичности кожи век, голубые склеры, миопия;
? уши: сверхрастяжимость;
? зубы: частичная адентия, сверхкомплектные зубы, опалесцирующая эмаль, пародонтоз, множественный кариес;
? характерным для этого заболевания является гипермобильность языка, так что больные легко достают языком кончик носа;
? грудная клетка: сколиоз, кифоз, лордоз, плоская спина, вдавление грудины;
? живот: грыжи (пупочная, белой линии, паховая, диафрагмальная), спонтанная перфорация кишечника;
? конечности: варикозные вены, подкожные подвижные узелки на голенях;
? сердце: пролапс митрального клапана, аритмии, вегетососудистая дистония;
? внутренние органы: птоз желудка, почек и матки; и мозг: аневризма сосудов мозга, субарахноидальное кровоизлияние;
? стремительные роды.
У 90% больных выявляется 1-3-й тип синдрома[8].
Клинические формы различаются между собой домированием того или иного симптомокомплекса.
В ходе общеклинического осмотра обращает на себя внимание сочетание у взрослых и детей синдрома гипермобильности и хронической боли, а также обобщенной гипералгезии. Наличие обобщенной гипералгезии может указывать на поражение центральной нервной системы в развитии хронической боли [9].
СЭД диагностируется по данным клинического обследования, изучения семейного анамнеза, и одного или более из ниже приведенных ДНК тестов[10].
2.3 Лабораторная диагностика
В стандартных общеклинических тестах специфических симптомов для СЭД, как правило, нет.
- Проявления геморрагического синдрома, которые часто встречаются у пациентов СЭД, обусловлены сосудисто-тромбоцитарным типом кровоточивости: тромбоцитопения на уровне субнормальных показателей, тромбоцитопатии.
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
- При разрыве магистральных сосудов, акушерских осложнений развивается смешанный тип кровоточивости. Анемия не типична, имеет преимущественно постгеморрагический характер.
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
- Рекомендовано проведение биохимических анализов, призванных выявить повышенное содержание различные метаболитов аномального коллагена: кислые аминогликаны, оксипролины; КФК и др.
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 4)
Источник: https://medi.ru/klinicheskie-rekomendatsii/sindrom-elersa-danlo-u-detej_14271/
Синдром Элерса-Данлоса: причины, виды, признаки, диагностика, как лечить

Синдром Элерса-Данлоса – врожденные патологические изменения соединительной ткани, обусловленные нарушением синтеза коллагеновых структур.
Эта группа наследственных коллагенопатий имеет системный характер и проявляется признаками поражения костно-суставной системы, кожи, кровеносных сосудов, органа зрения.
Основными симптомами патологии являются: излишняя гибкость суставов, перерастяжение кожи, слабость сосудистой стенки, поражение костей позвоночника, патология зрительного анализатора.
Синдром Элерса-Данлоса был назван в честь своих первооткрывателей – дерматологов из Дании и Франции Э. Элерса и А. Данлоса. Они первые описали причины и симптомы данной патологии.
В настоящее время существует 6 основных форм синдрома Элерса-Данлоса, которые отличаются симптоматикой, способом наследования, биологическими и химическими изменениями в тканях.
Среди всех существующих типов дисплазий этот синдром считается самым распространенным.
примеры людей с синдромом Элерса-Данлоса
Диагностика патологии основывается на симптоматике, результатах гистологического и генеалогического исследований. Данное заболевание является неизлечимым.
Больным назначают лекарственные препараты, уменьшающие выраженность основных симптомов.
В настоящее время синдром Элерса-Данлоса интересует врачей различных медицинских специальностей: генетиков, травматологов-ортопедов, дерматологов, терапевтов, окулистов.
Классификация
Классическая или традиционная классификация синдрома Элерса-Данлоса:
- I и II тип — классический,
- III тип — гиперподвижный,
- IV тип — сосудистый,
- V тип – связанный с Х хромосомой,
- VI тип – кифосколиоз,
- VII тип — артрохалазия,
- VIII тип – периодонтит,
- IX тип – дерматоспараксис,
- X тип – полосовидная атрофия кожи,
- XI тип – семейная нестабильность суставов.
Первая традиционная классификация синдрома Элерса-Данлоса включала более 10 типов заболевания. В 1997 году ее упростили и выделили 6 типов патологии. В современную классификацию не вошли редко встречающиеся формы, которые регистрировались у членов исключительно одной семьи или были недостаточно охарактеризованы.
6 типов на которые в настоящее время подразделяют синдром Элерса-Данлоса:
- Гиперподвижный,
- Классический,
- Сосудистый,
- Кифосколиоз,
- Артроскалазия,
- Дерматоспараксис.
Этиология
Этиология и патогенез синдрома Элерса-Данлоса в настоящее время до конца не изучены. Известно, что единственной причиной патологии является мутация генов, отвечающих за выработку коллагена, являющегося важной частью соединительной ткани и позволяющего ей растягиваться.
Дефицит коллагеновых волокон приводит к качественной и количественной соединительнотканной патологии. В организме больного снижается активность целого ряда ферментов, возникает недостаточность аминокислот, входящих в состав коллагенового волокна.
При этом истончается дерма, теряется компактность коллагеновых волокон, разрастаются эластические волокна, увеличивается число сосудов, расширяется их просвет.
Нарушение выработки коллагена обусловлено генетическими аномалиями. В результате мутации генов изменяются или полностью утрачиваются его функции.Аминокислоты выпадают из полипептидной цепочки, изменяется форма тройной спирали, нарушаются свойства тканей и органов.
Эластические волокна гипертрофируются и перекручиваются, коллагеновые волокна разрушаются, что приводит к мукоидному отеку соединительной ткани — первой стадии ее дезорганизации.
Группу риска составляют все члены семьи, в которой имелись случаи возникновения данного заболевания.
Симптоматика
Полиморфизм клинических признаков патологии объясняется присутствием соединительной ткани во всех органах и системах человеческого организма. Симптомы заболевания у разных людей могут иметь различную степень выраженности: у одних быть незначительными, а у других – серьезными, угрожающими жизни.
В первую очередь патологическим изменениям подвергаются кожа и суставы, что связано с максимальным содержанием в них коллагена.
- Кожный синдром – ведущий клинический признак патологии. У больных кожа становится гиперэластичной, слишком мягкой и хрупкой. Она легко собирается в складку и оттягивается. Нежная и бархатистая кожа морщинится на ладонях и стопах. На ней появляются темно-коричневые пятна, множество келоидных рубцов, просвечивающиеся вены, стрии на пояснице. Подобные признаки максимально выражены у новорожденных детей. Минимальная травматизация хрупкой и ранимой кожи приводит к образованию ран, на месте которых формируются грубые рубцы. Легкие порезы часто перерастают в серьезные, медленно и плохо заживающие раны. Во время хирургических вмешательств такая кожа плохо поддается сшиванию: она лопается и рвется. Рубцы на месте ран со временем покрываются грубыми наростами.
- Суставные проявления – гипермобильность суставов локального или распространенного характера. Суставы становятся свободными, сильно гнущимися. У больных опухают и растягиваются связки, часто возникают вывихи, сопровождающиеся сильной болью. Суставные проявления становятся впервые заметными, когда маленький ребенок начинает ходить. По мере роста и развития детского организма этот клинический признак становится менее заметным.
- Поражение сосудистой системы проявляется ломкостью и хрупкостью капилляров, вен и артерий, а также крупных сосудов — аорты и легочного ствола. Кровеносные сосуды у больных разрываются, появляются синяки и кровотечения. У некоторых пациентов поражаются сердечные клапаны, начинают кровоточить десна.
- Глазные симптомы — близорукость, хрупкость склеры, отверстия в глазном яблоке, опущение верхнего века, эпикантус – складка кожи по обе стороны от носа, охватывающая внутренний угол глаза.
- Поражение скелета — грудная клетка приобретает форму воронки, развивается плоская спина, лордоз, сколиоз или кифоз, а также их комбинированные варианты, косолапость, прикус становится неправильным, зубы растут неровными и выпадают. Поражение зубов проявляется воспалением околозубной ткани, адентией, кариесом.
- Отмечается также поражение дыхательной системы, желудочно-кишечного тракта, женских половых органов.
Симптоматика основных типов
- Гиперподвижный тип проявляется нестабильностью и чрезмерной мобильностью суставов. Больной легко сгибает мизинец на 90 градусов и переразгибает коленный и локтевой сустав на 10 градусов и более. У пациентов обнаруживают плоскостопие, «готическое» небо, сколиоз или кифоз, неровные зубы.
Их кожа бархатная и гладкая, склонна к повреждениям и кровоподтекам. У женщин в климактерическом периоде снижается плотность костей. Поражение сердца и сосудов проявляется ортостатической тахикардией, признаками пролапса митрального клапана. У больных развиваются дисфункции кишечника, появляются признаки сдавления нервов — парестезии, боли.
Пациенты на приеме у врача жалуются на мышечные и суставные боли, чрезмерную усталость, апатию. Опорно-двигательная боль при данном типе патологии становится просто изнурительной. Болевой синдром требует регулярного приема обезболивающих препаратов на протяжении всей жизни.
- Классический тип проявляется разболтанностью суставов, патологическим растяжением кожи, большим количеством шрамов. Чтобы заболевание появилось, достаточно унаследовать мутировавший ген от одного из родителей. Грубые рубцы с блестящей лоснящейся поверхностью располагаются в наиболее уязвимых местах — на коленках, локтях, лбу.
Под бархатистой кожей определяется множество кистозных и опухолевых образований. Мыщечная слабость приводит к появлению характерных грыж, опущению соматических органов.
- Сосудистый тип — появление на бледной и даже синюшной коже крупных гематом, экхимозов и ран. Сосудистый тип возникает редко, но протекает тяжело.
Наследуется он различными путями – доминантным или рецессивным. У больных повышенной подвижностью обладают только суставы кисти. Через тонкую и бледную кожу просвечиваются подкожные сосуды. Геморрагический синдром — основной клинический признак заболевания. Гематомы, разрывы сосудов и полых органов обуславливают высокую летальность патологии.
Это очень опасная форма, приводящая к смерти больного. Изменения геморрагического характера наблюдаются у лиц с нормальным коагуляционным статусом. Эту особенность заболевания необходимо учитывать хирургам при проведении операций.
- Кифосколиозный тип сопровождается выраженными нарушениями осанки и характеризуется общей гипермобильностью суставов, слабостью мышц, нарушением моторного развития, искривлением позвоночника — кифозом или сколиозом, хрупкостью сосудов, частым появлением ран на коже, грубых шрамов и гематом, беспричинной отслойкой сетчатки.
- Артрохалазия проявляется повышенной мобильностью суставов, гиперрастяжимостью кожи и маленьким ростом больного.
- При дерматоспараксисе структура кожи становится мягкой и рыхлой, на ней периодически появляются гематомы. Она отслаивается пластами и приобретает тестообразную консистенцию. Заживление ран происходит без формирования атрофии. У некоторых больных появляются большие грыжи.
: ЛФК при синдроме Элерса-Данлоса
Прогноз патологии в большинстве случаев довольно благоприятный. Синдром Элерса-Данлоса существенно снижает качество жизни больных, но практически не влияет на ее продолжительность. Дети, рожденные с данным заболеванием, часто имеют врожденные заболевания внутренних органов и пороки сердца. Им оформляется пожизненная инвалидность.
Медико-генетическое консультирование — основа профилактики данного заболевания. Его проводят будущим родителям в тех семьях, где регистрировались случаи синдрома.
Правильный выбор профессии, щадящий режим физической активности и соблюдение всех рекомендаций специалистов помогут существенно снизить риск развития тяжелых осложнений. Больным лицам запрещены контактные виды спорта и противопоказана мануальная терапия.
Гиперподвижные суставы необходимо защищать и стабилизировать с помощью бандажей и фиксаторов.
Во время занятий спортом необходимо пользоваться специальными индивидуальными защитными средствами – различными видами касок, шлемов, очков, перчаток, наколенников, налокотников, берушей, поясов. Больным следует ежегодно проходить ЭКГ, посещать офтальмолога и кардиолога. При необходимости им проводят курсовое кардиотрофическое лечение.
Синдром Элерса-Данлоса — тяжелая патология, опасность которой
зависит от отдельной мутации. Чрезмерно эластичная и тонкая кожа больных легко повреждается, образовавшиеся раны заживают долго и неправильно, суставы излишне гнутся. В случае выявленного первичного биохимического дефекта необходимо проведение пренатальной диагностики.
: люди с синдромом Элерса-Данлоса о своем заболевании
Источник: https://sindrom.info/elersa-danlosa/